
Air pollution is a complex mixture of gases (e.g., ozone), particulate matter, and organic compounds present in outdoor and indoor air. Dogs exposed to severe air pollution exhibit chronic inflammation and acceleration of Alzheimer’s-like pathology, suggesting that the brain is adversely affected by pollutants. We investigated whether residency in cities with high levels of air pollution is associated with human brain inflammation. Expression of cyclooxygenase-2 (COX2), an inflammatory mediator, and accumulation of the 42-amino acid form of β-amyloid (Aβ42), a cause of neuronal dysfunction, were measured in autopsy brain tissues of cognitively and neurologically intact lifelong residents of cities having low (n:9) or high (n:10) levels of air pollution. Genomic DNA apurinic/apyrimidinic sites, nuclear factor-κB activation and apolipoprotein E genotype were also evaluated. Residents of cities with severe air pollution had significantly higher COX2 expression in frontal cortex and hippocampus and greater neuronal and astrocytic accumulation of Aβ42 compared to residents in low air pollution cities. Increased COX2 expression and Aβ42 accumulation were also observed in the olfactory bulb. These findings suggest that exposure to severe air pollution is associated with brain inflammation and Aβ42 accumulation, two causes of neuronal dysfunction that precede the appearance of neuritic plaques and neurofibrillary tangles, hallmarks of Alzheimer’s disease.
INTRODUCTION
Air pollution is a complex mixture of gases (e.g., ozone), particulate matter (PM),1 and organic compounds present in outdoor and indoor air. Controlled exposures to ozone and PM cause respiratory tract injury and inflammation, as well as increased levels of mediators of inflammation circulating in the blood (American Thoracic Society, 1996; van Eeden and Hogg, 2002), while chronic exposure to air pollution is associated with respiratory and cardiovascular-related sickness and death (Brunekreef and Holgate, 2002). Children in Mexico City, a metropolitan area with ozone and PM levels that consistently exceed the U.S. air quality standards, exhibit evidence of chronic inflammation of the upper and lower respiratory tracts, alterations in circulating inflammatory mediators and breakdown of the respiratory epithelial barrier in the nose (Calderon-Garciduenas et al., 2003b). Likewise, dogs residing in Mexico City experience chronic upper and lower respiratory tract inflammation and breakdown of both the respiratory and olfactory epithelial barriers (Calder´onGarcidue˜nas et al., 2001, 2002, 2003a). The brains of these dogs exhibit upregulation of COX2 expression, indicative of chronic brain inflammation, and accelerated accumulation of Aβ42, a neurotoxic fragment of the amyloid precursor protein (APP) that causes neuronal dysfunction (Selkoe, 2001; Calderon-Garciduenas et al., 2003a). The canine data suggested that chronic exposure of humans to severe urban air pollution may also have adverse effects in human brains.
MATERIALS AND METHODS
Study Cities
We conducted a study using autopsy brain samples from Mexican subjects, lifelong residents of 2 large cities with severe air pollution, Mexico City and Monterrey, and 5 small cities with low levels of air pollution, Abasolo, Iguala, El Mante, Tlaxcala, and Veracruz. Mexico City (MC) is a megacity with 20 million inhabitants, 3.5 million vehicles and extensive industrial activity (Bravo and Torres, 2002). Monterrey is the second largest industrial city in the country with 3.5 million residents and thousands of industries (Junco-Munoz et al., 1996). Ozone (O3) and PM are the major air pollutants for both MC and Monterrey. Bacterial lipopolysaccharides (LPS) and metals are important components of PM (Junco-Munoz et al., 1996; Bravo and Torres, 2002; Osornio-Vargas et al., 2003). The climatic conditions are remarkably stable in both cities, so pollutant concentrations are consistent from year-to-year, while ozone and PM levels exceed the U.S. standards most of the year (Junco-Munoz et al., 1996: Bravo and Torres, 2002). In the cities with low levels of air pollution, the combination of relatively few emission sources from industry and cars, and good ventilation conditions by regional winds, keep criteria pollutant concentrations below the current U.S. standards. Three additional factors considered in the selection of the control cities included: (1) an altitude above sea level similar to Mexico City, (2) studies in dogs from these cities showing minimal pathology in lungs and hearts (Calderon-Garciduenas et al., 2001), and (3) knowledge that children residing in control cities show no evidence of upper or lower respiratory pathology and no systemic involvement (Calderon-Garciduenas et al., 2003b).
Autopsy Selection
The study protocol was approved by the Institutional Review Boards for Human Studies at the Institutions involved in Mexico. Data available for all subjects included age, gender, place of birth, place of residency, occupation, years of completed education, smoking habits, medications, clinical histories, nutritional status, cause of death, and time between death and autopsy. We studied 19 subjects, who came to autopsy from the selected cities and fulfilled the inclusion criteria. The low-exposure group included 9 subjects, and the high-exposure group included 10 subjects, 5 from Mexico City and 5 from Monterrey. All subjects were nonsmokers of middle socioeconomic status. In addition to nonsmoking status, inclusion criteria were: (1) access to recent complete clinical information within 3 months of death through medical records, interviews with private practitioners, and evaluation of job performance; (2) no evidence of neurological disease or cognitive abnormalities by medical history, and by complete neuropathological examination; (3) body mass index between 19 and 25; (4) negative family history of dementia; (5) negative history of drug addictions, occupational exposures to potential neurotoxicants, recent vaccinations, and intake of vitamins, dietary or herbal supplements; and a (6) negative history of nonsteroidal anti-inflammatory drugs (NSAID) and steroidal compounds.
Cause of death was considered for all subjects to rule out the possibility that infection, inflammatory events, brain ischemia, or hypoxia might impact gene expression levels measured in the study. Based on evaluation of the clinical medical records and information obtained from relatives and coworkers by 2 physicians, each subject was considered cognitively and neurologically intact. The subjects had no clinical history or pathological evidence of short- or long-term inflammatory processes, administration of anti-inflammatory drugs or hormones, or events such as cerebral ischemia, head trauma, or epilepsy. A negative history of depression, memory complaints, and speech or learning disabilities was required.
Autopsy and Tissue Preparation
Autopsies were performed 4.1 ± 1.3 hours after death. The postmortem period was similar for controls and exposed. The skull was opened and the olfactory bulbs and the brain removed. Olfactory bulbs and selected areas from alternating right and left cerebral hemispheres were immediately frozen in liquid nitrogen and kept at −80◦C. Frozen tissue for the RT-PCR and AP sites was taken from the cortex and the white matter, taking care to make a perpendicular cut to the brain surface and keeping similar amounts of cortex and white matter for each method. Sections adjacent to the frozen material were immersed in 10% neutral formaldehyde, fixed for 48 hours, and transferred to 70% alcohol. Sections were taken from superior frontal gyrus, hippocampus, and olfactory bulbs. Lung sections were also taken. Paraffin sections 6 µm thick were cut and stained with hematoxylin and eosin. Immunohistochemistry (IHC) was performed using C-terminal specific Aβ42 antibody from BD Transduction Laboratories (San Diego, CA); NF-κB p65 from Santa Cruz Biotechnology (Santa Cruz, CA); glial fibrillary acidic protein (GFAP) antibody from Roche Applied Science (Indianapolis, IN), and a human COX2 polyclonal antibody from Cayman Chemical Company (Ann Arbor, MI). Negative controls included omission or substitution of primary antibodies by nonspecific, isotype-matched antibodies. Quantitative analysis of COX2 and Aβ42 was performed with the BioQuant Nova prime image analysis system (Bioquant Image Analysis Corp, Nashville, TN). To quantify the number of AP sites we used an aldehyde reactive probe (ARP) (Calderon-Garciduenas et al., 2003a). AP sites are expressed per 1 × 106 nucleotides.
Tissue selection and preservation were performed by neuropathologists using uniform procedures. The immmunohistochemical and biochemical procedures were performed at the University of North Carolina. Evaluation of samples and quantification of gene expression were done blindly by experienced researchers with no access to the codes regarding the clinical characteristics or the geographical origin of the subjects.
Real-Time RT-PCR
Messenger RNA abundances were measured by real-time RT-PCR analysis of total RNA isolated from the frontal cortex or hippocampus using Trizol reagent (InVitrogen, San Diego, CA). Random-primed first-strand cDNAs were generated as described (Carson et al., 2002) except that 1 microgram of total RNA was used as template. Relative abundances of mRNAs were estimated by quantitative fluorogenic 5′ nuclease (TaqMan) assay of the first strand cDNAs as described (Carson et al., 2002). Primers and fluorophore-labeled TaqMan probes targeting human COX2, APP695, and APP751 were designed using Primer Express software (Applied Biosystems, Foster City, CA) or Primer Designer software (Scientific and Educational Software, Durham, NC) based upon sequence information in GenBank. Probes were labeled at the 5′ end with 6-carboxy-fluorescein (FAM) or 6-carboxy-4, 5-dichloro-2,7-dimethoxyfluorescein (JOE) and at the 3′ end with 6-carboxytetramethylrhodamine (TAMRA) or Black Hole Quencher-1 (BHQ1). Primer and probe sequences were as follows: COX2, sense, 5′ -GAATCATTCACCAGGCAAATTG-3 , antisense, 5′ -TCTGTACTGCGGGTGGAACA-3′ , probe, 5′ -FAM-TCCTACCACCAGCAACCCTGCCA-TAMRA-3′ ; APP695 sense, 5′ -GGTGGTTCGAGTTCCTACAA-3′ ; APP751 sense, 5,- GCAGCGCCATTCCTACA-3′ ; APP695 and APP751 common antisense, 5′ -TGGAAATGGGCATGTTCATTCT-3′ , and APP695 and APP751 common probe, 5′ -JOE-CAGCCAGTACCCCTGATGCCGT-BHQ1-3′ . To control for unintentional sample to sample variation in (1) the amount of total RNA reverse transcribed and (2) reverse transcription efficiency, the amount of COX2, APP695, and APP751 mRNA in each sample was normalized to the amount of 18s ribosomal RNA (rRNA), yielding an index (molecules per femtomol 18s rRNA) proportional to the relative abundance of the mRNA in each sample. The amount of 18s rRNA in each first-strand cDNA was estimated using TaqMan ribosomal control reagents (Applied Biosystems, Foster City, CA) and serial dilutions of a recombinant plasmid standard.
Apolipoprotein E Genotyping
DNA was isolated from frontal cortex as described and genotyped for the HhaI restriction site polymorphism in the ApoE gene (Hixson and Vernier, 1990).
Statistical Analysis
Statistics were performed using Stata Statistical software (College Station, TX). We applied the t-parametric procedure that considers the differences among variances of the variable of interest, COX2 mRNA abundance, in the low- and high-exposure subjects. Then, we considered the transformation of the response variable to stabilize the variances in our model that considers covariables such as age, gender, and APP isoform mRNA abundances in frontal cortex and hippocampus analyzed independently. Pearson and Spearman correlations were also calculated for all the variables in the model, covariables and the response variable. The quantification of COX2 mRNA abundance by RT-PCR and COX2 and Aβ42 IHC by image analysis was analyzed by the Mann–Whitney U-test. Significance was assumed at p < 0.05. Data are expressed as mean ± SEM. The influence of individual data points on linear regressions was examined using leverage-residual (L-R) plots and the DFFITS statistic.
RESULTS
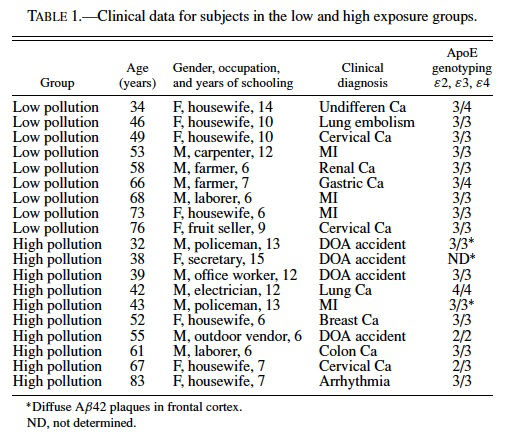
The average age and years of schooling for low- and high-exposure groups were 58.1 ± 4.6 and 51.2 ± 4.9 years of age and 9.2 ± 0.86 and 9.7 ± 1.1 years of schooling, respectively. The primary causes of death included accidents resulting in immediate death, arrhythmias, myocardial infarction, and carcinomas including: gastric, lung, colon, breast, and cervical. Clinical data for control and exposed subjects is presented in Table 1.
Evidence of chronic respiratory tract inflammation was present in residents of cities with severe air pollution. Lungs displayed chronic mononuclear cell infiltrates along with macrophages filled with PM surrounding the bronchiolar walls, and extending into adjacent vascular structures. Peribronchial lymph nodes exhibit abundant PM. In contrast, subjects from control cities exhibited rare foci of inflammatory cells in association with terminal bronchioles and peribronchial lymph nodes showed small clusters of PMcontaining macrophages.
Gross and routine microscopic brain examinations were unremarkable in all subjects.
We selected COX2 mRNA levels as the primary variable because of the striking upregulation of COX2 expression observed in the brains of dogs residing in Mexico City (Calderon-Garciduenas et al., 2003a) and because enhanced neuronal COX2 expression in hippocampus is a feature of the clinical progression of early Alzheimer’s disease (Ho et al., 2001). COX2 mRNA abundance was measured by real-time RT-PCR analysis of total RNA isolated from frontal cortex and hippocampus. In the 19 subjects for which frontal cortex tissue was available, there was an elevation of COX2 mRNA levels in the high exposure group (p = 0.009, Mann–Whitney test) (Figure 1A). An elevation of COX2 immunoreactivity in subjects of the high-exposure group was confirmed by quantitative image analysis of COX2 immunoreactivity (IR) (p = 0.01, Mann–Whitney test) (Figure 1B). In subjects from the low-exposure group, COX2 IR was confined to neuronal cell bodies, whereas subjects from the high exposure group exhibited COX2 staining in neuronal cell bodies and dendrites, as well as strong COX2 staining of the endothelium both in the cortex (Figure 1C) and white matter blood vessels.
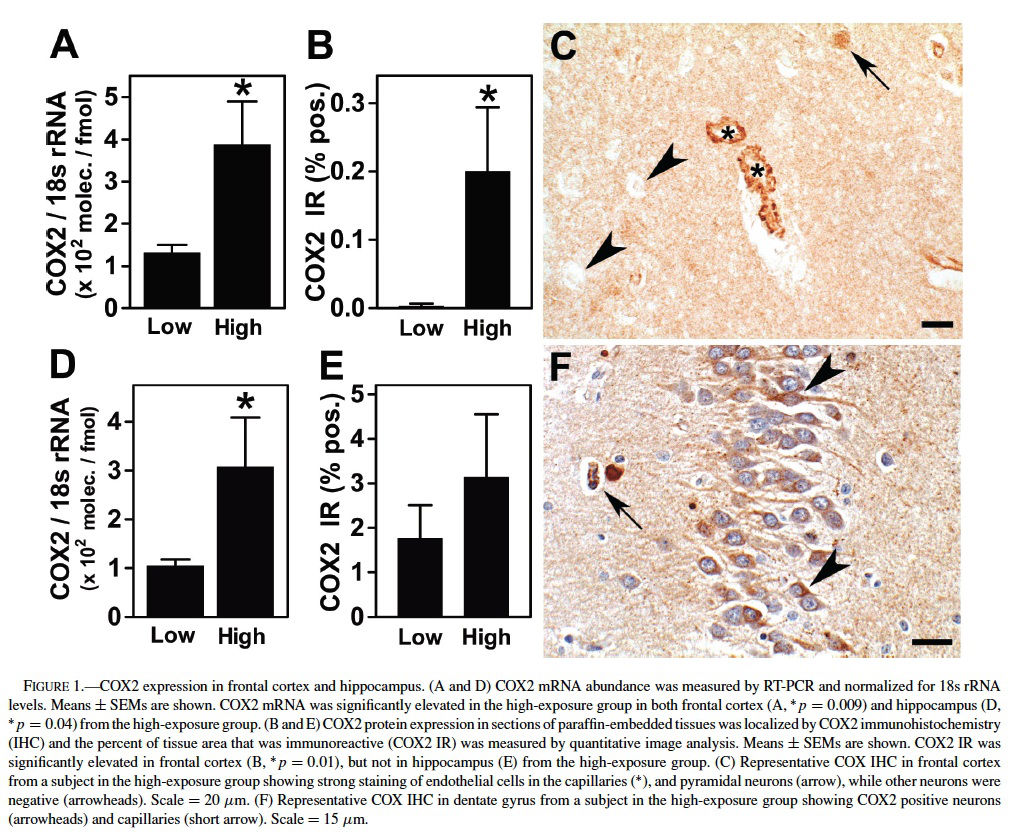
In the 15 subjects for which hippocampus tissue was available, COX2 mRNA was also elevated in the high exposure group (p = 0.045, Mann–Whitney test) (Figure 1D). Although the average density of COX2 IR was greater in the high-exposure group, it was not significantly different from levels in the low-exposure group (p = 0.37) (Figure 1E). Hippocampal COX2 IR in highly exposed subjects was observed in neuronal bodies included in the CA1-2 and CA4 regions and the dentate gyrus, as well as endothelial cells (Figure 1F), whereas control subjects had neuronal staining, but no endothelial staining.
Spearman and Pearson correlations showed a strong positive association between COX2 mRNA levels and oxidative DNA damage as measured by AP sites (r = 0.89, p = 0.001, Spearman and Pearson) in frontal cortex of the high-exposure group, but not the low exposure group (Figure 2). There was no correlation of COX2 mRNA levels and AP sites in hippocampus.

The upregulation of COX2 expression is a common feature of inflammatory responses (Cao et al., 1997; Lukiw and Bazan, 1997; Yermakova and O’Banion, 2001), suggesting that the increase of COX2 in the high-exposure group is the result of a chronic inflammatory process. Inflammatory responses are also characterized by activation of nuclear factor-κB (NF-κB), an inducible transcription factor that regulates the expression of a variety of genes whose products mediate inflammatory and immune responses (Mattson and Camandola, 2001). Nuclear localization of NF-κB IR, an indication of NF-κB activation, was seen in frontal cortex neurons and glial cells of subjects from the high-exposure group. There was no indication of NF-κB activation in endothelium. Nuclear localization of NF-κB was not observed in the frontal cortex of subjects from the low exposure group.
Aβ42 accumulation was examined in frontal cortex and hippocampus because upregulation of COX2 expression in dogs was associated with Aβ42 accumulation (Calderon-Garciduenas et al., 2003a), and because of reports of significant correlations between elevated levels of Aβ42 and cognitive decline (Naslund et al., 2000). In subjects from the high-exposure group, Aβ42 selectively accumulated in the perikaryon of pyramidal frontal neurons and was present in cortical and white matter astrocytes, and subarachnoid and cortical blood vessels (Figure 3A and B). Quantitative estimation of Aβ42 IR confirmed an increase in Aβ42 accumulation in the frontal cortex (p = 0.04, Mann–Whitney test) (Figure 3C, and in hippocampus (p = 0.001, Mann–Whitney test) (Figure 3D) of the high exposure group. Rare diffuse Aβ42 plaque-like staining was present in the frontal cortex of 3 subjects in the high exposure group (32, 38, and 43 years old). The diffuse Aβ42 plaques were associated with reactive astrocytes (Figure 3E) or apoptotic nuclei (not shown). The diffuse plaque-like staining was not associated with the apolipoprotein E ε4 allele (see Table 1), a risk factor for the development of Alzheimer’s disease (Selkoe, 2001).

The predominant isoform of the amyloid precursor protein (APP) in brain consists of 695 amino acids (APP695). Expression of an alternatively spliced isoform consisting of 751 amino acids (APP751), including a Kunitz protease inhibitor domain, increases following ischemia and neurotoxic damage in the aged brain and in AD (Sandbrink et al., 1994; Moir et al., 1998). We measured APP695 and APP751 mRNA abundances in frontal cortex. Neither APP751 mRNA levels nor the ratio of APP751 to APP695 mRNA levels differed significantly between the high- and low-exposure groups. However, COX2 mRNA levels in frontal cortex from MC and Monterrey subjects were positively associated with APP751 mRNA expression levels (p = 0.05, Spearman) and APP695 mRNA levels (p = 0.05, Spearman). APP751 mRNA was strongly upregulated in 2 of the 3 subjects from the high-exposure group with Aβ42 diffuse plaques (4.3- and 5.2-fold higher compared to high-exposure subjects without plaques). These data suggested that an increase in APP751 mRNA expression is a feature of the high-exposure group, but that the average increase is small compared to the average expression levels in the low-exposure group.
As an exploratory exercise, olfactory bulbs were obtained from a limited number of subjects. COX2 and Aβ42 immunoreactivity were present in the olfactory bulb of all 3 subjects in the high exposure group for whom olfactory bulb tissue was available. These subjects corresponded to those who had rare Aβ42 plaque-like staining present in the frontal cortex. Aβ42 accumulated in mitral and tufted neurons and ensheathing cells (Figure 4A). Aβ42 immunoreactivity was also present in ensheathing cells and astrocytes of olfactory nerves in the same subjects (Figure 4B). COX2 and Aβ42 staining were not present in the 2 subjects from the low exposure group for which olfactory bulb and nerve were examined.
DISCUSSION
The residents of the 2 highly polluted cities included in this study were chronically exposed to a complex mixture of air pollutants, including ozone and PM. Ozone causes respiratory tract epithelial injury and inflammation (American Thoracic Society, 1996), while PM contains bacterial lipopolysaccharide (LPS) and combustion-derived metals such as nickel and vanadium—all agents that can evoke inflammatory responses (Elmquist et al., 1997; Calderon-Garciduenas et al., 2003a; Osornio-Vargas et al., 2003). Ultrafine PM (PM with mean aerodynamic diameter <100 nm), when deposited in the lung, causes severe inflammation (Oberdorster, 2001). Previous studies have shown that human and dog residents of Mexico City have a sustained inflammatory process in the upper and lower respiratory tract (Calderon-Garciduenas et al., 2001, 2003b) and apparently healthy children exhibit an imbalance of serum cytokine levels (Calderon-Garciduenas et al., 2003b).
This and previous studies have provided evidence of brain inflammation in the form of an upregulation of COX2 expression in the brains of both human and dog residents of Mexico City (Calderon-Garciduenas et al., 2003a). COX2 expression was predominantly observed in capillary endothelium. Brain blood vessels exhibit constitutive and induced expression of receptors for TNF-α, IL-1β, and IL-6 (Rivest, 2001; Nguyen et al., 2002). Chronic respiratory tract inflammation may lead to chronic brain inflammation by altering levels of circulating cytokines, such as TNF-α and IL-1β that have the ability to evoke expression of inflammatory mediator genes, such as COX2, within brain capillary endothelium (Rivest, 2001) and that are known to be transported across the blood–brain barrier (Rivest, 2001; Nguyen et al., 2002).
The increase in COX2 expression in brain endothelium was not associated with an increase in endothelial nuclear NF-κB in this study nor in dog residents of MC (Calderon-Garciduenas et al., 2003a). This is noteworthy because acute increases in the expression of COX2 evoked by proinflammatory cytokines such as IL-1β and TNF-α are mediated in part by the acute activation of NF-κB, which is often indicated by an increase in nuclear NF-κB. However, COX2 expression is also regulated by control of the stability and translation of its messenger RNA, processes that are NF-κB-independent (Dixon et al., 2001). Furthermore, the activity of nuclear NF-κB can increase in the absence of increased nuclear NF-κB abundance (Finco et al., 1997). The discordance between increased endothelial COX2 expression and nuclear NF-κB may reflect distinct mechanisms of the regulation of COX2 expression in acute versus chronic conditions.
We observed both an upregulation of COX2 expression and the accumulation of Aβ42 in the high-exposure group. Aβ42 accumulation may be a consequence of the increase in COX2 expression. Neuron-specific ectopic expression of human COX2 in the brain of a mouse model of Alzheimer’s disease increased β-amyloid plaque formation and elevated Aβ40 and Aβ42 with no change in total APP, suggesting that COX2 influences APP processing (Xiang et al., 2002). Using an adenoviral gene transfer system to study the effects of COX1 and COX2 on β-amyloid generation, Qin and coworkers (2003) found that COXs promote Aβ generation via a PG-E2-mediated activation of γ -secretase, the membrane proteinase that generates Aβ40 and Aβ42. These findings suggest that upregulation of COX2 expression may accelerate Aβ42 accumulation.
Recent studies showing that nonsteroidal anti-inflammatory drugs (NSAIDs) that do not inhibit COX2 activity do inhibit Aβ42 accumulation, suggest that COX2 may act on Aβ42 accumulation through an NSAID-sensitive intermediate. Aβ42 production in vivo and in vitro may be regulated by RhoA GTPase and its associated kinases, which appear to be the targets of NSAIDs that lower Aβ42 accumulation (Zhou et al., 2003). Rho-dependent signaling may play a role in the upregulation of COX2 expression as well (Aznar et al., 2003). If this is the case, the ability of NSAIDs that inhibit RhoA GTPase and COX2 expression might be effective at ameliorating the neuropathology reported here.
COX2 is an extremely potent biologically active mediator of inflammation, and the first rate-limiting enzyme in the production of prostacyclins, prostaglandins, and thromboxane from arachidonic acid. The positive correlation we observed between COX2 mRNA and AP sites in frontal cortex may be a consequence of COX2-mediated prostanoid synthesis, a major source of reactive oxygen species that are capable of damaging DNA.
LPS is a component of PM, including Mexico City PM (Osornio-Vargas et al., 2003). Systemic administration of LPS to rodents causes upregulation of COX2 in brain perivascular and endothelial cells (Elmquist et al., 1997; Yermakova and O’Banion, 2001) and alters the expression or processing of APP or both. It also accelerates the generation of Aβ42 in transgenic mice that overexpress the Swedish variant of APP695 (Sheng et al., 2003) and increases the abundance of APP751 (Brugg et al., 1995). LPS could cause these effects by several mechanisms. It could evoke a local innate immune response in the respiratory tract leading to altered systemic levels of cytokines that can act on the brain microvasculature. LPS-induced cytokines or LPS itself could elicit neurogenic inflammation by activating sensory nerves in the respiratory tract such as the vagus (Elmquist et al., 1997; Roth and De Souza, 2001). In addition, Nguyen and coworkers (2002) suggest that LPS in the systemic circulation could bind its transmembrane receptors on microglia residing in areas devoid of blood–brain barrier.
Previous studies have shown that residents of Mexico City suffer breakdown of their nasal respiratory epithelial barrier (Calderon-Garciduenas et al., 2003b). Breakdown of the nasal olfactory epithelial (nose-brain) barrier is also likely in Mexico City residents, because this has been observed in dogs (Calderon-Garciduenas et al., 2002, 2003a). Breakdown of respiratory epithelial barriers may contribute to brain inflammation by increasing the access of air pollutants to the brain, both directly through the olfactory pathway and indirectly through the systemic circulation (Calderon-Garciduenas et al., 2002; Dorman et al., 2002; Calderon-Garciduenas et al., 2003a; Oberdorster et al., 2004). The olfactory bulb is the first synaptic relay of neurons that reside in the olfactory epithelium and that are directly exposed to air pollutants. Olfactory neurons are capable of taking up and transporting environmental agents into the brain (Dorman et al., 2002). Oberdorster and coworkers (2004) have shown recently that inhaled ultrafine PM accumulates in rat olfactory bulb, suggesting that olfactory neurons take-up and transport ultrafine PM. The presence of olfactory bulb pathology in subjects from the high-exposure group suggests that the human nose may be a portal of entry of air pollutants into the brain as well.
The pathology observed in the subjects chronically exposed to severe air pollution has a number of similarities to the pathology of Alzheimer’s disease (AD). Both COX2 expression and oxidative DNA damage are elevated in the early stages of Alzheimer’s disease (Ho et al., 2001; Nunomura et al., 2001). Increased abundance of APP751 such as that observed in 2 of the high-exposure subjects is a characteristic of Alzheimer’s brain (Selkoe, 2001) and olfactory bulb pathology similar to that observed in a small number of individuals in the high-exposure group is one of the earliest pathological findings in Alzheimer’s disease (Braak et al., 1998), where it is associated with impaired olfaction.
We did not observe neurofibrillary tangles or Aβ neuritic plaques, which are characteristic of a histopathological diagnosis of AD. This is not surprising given the relatively young average age of the subjects. However, the absence of neurofibrillary tangles or Aβ plaques does not imply an absence of AD pathogenesis, a process that takes decades and that currently has no indisputable histopathological characteristics. Recently it has been proposed that an increase in soluble Aβ42 is the causative agent of AD (Selkoe, 2001). Indeed, intraneuronal Aβ42 accumulation in target areas precedes Aβ plaque deposition and neurofibrillary tangles in AD (Gouras et al., 2000; Selkoe, 2001; Gyure et al., 2001). The Aβ42 accumulation observed in subjects from the high-exposure group may be an indication of AD pathogenesis preceding the development of neurofibrillary tangles and Aβ plaques.
Taken together, the histochemical and biochemical changes observed in subjects from the high-exposure group suggest that exposure to urban air pollution may cause brain inflammation and accelerate the accumulation of Aβ42, a putative mediator of neurodegeneration and AD pathogenesis. However, this conjecture is supported in part by APP751 mRNA expression and olfactory bulb pathology in 3 individuals in the high-exposure group. It is possible that these individuals are unusually susceptible to air pollution. On the other hand, this association may be coincidental. For example, these individuals may have genetic susceptibilities to AD that are unrecognized. Additional studies involving a larger number of subjects are needed to address this limitation.
Currently, Alzheimer’s disease is an irreversible, fatal brain disorder that diminishes the quality of life of affected individuals and places a burden on the health care system (Hebert et al., 2003). In 2000 there were 4.5 million people with AD in the United States and it is projected that AD will affect 13.2 million by 2050 (Hebert et al., 2003). The role played by the environment in the pathogenesis of AD is unknown. The identification and mitigation of environmental factors that influence AD pathogenesis is one approach to limiting the future impact of AD. About half of all Americans live in areas where levels of smog are unhealthy. Furthermore, significant numbers of individuals suffer occupational and indoor exposures to air pollutants. Our findings suggest that these exposures may accelerate the appearance of precursors of Alzheimer’s disease.
While we cannot exclude with certainty the possibility that the neuropathology described in highly exposed individuals is the result of conditions present in a large, urban environment other than chronic exposure to air pollutants, this research has identified a potential public health risk of great importance. The findings suggest a clear need for epidemiological and toxicological studies that can more fully characterize the association between chronic exposure to air pollutants and the risk of developing Alzheimer’s disease.
ACKNOWLEDGMENTS
We deeply appreciate the assistance of Robert Schoonhoven and Jennifer Dulyx from the University of North Carolina at Chapel Hill with immunohistochemistry.
REFERENCES
American Thoracic Society (1996). Health effects of outdoor air pollution. Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society. Am J Respir Crit Care Med 153, 3–50.
Aznar, B. S., Valer´on, P. F., and Lacal, J. C. (2003). ROCK and nuclear factor- κB-dependent activation of cyclooxygenase-2 by Rho GTPases: effects on tumor growth and therapeutic consequences. Mol Biol Cell 14, 3041– 54.
Braak, H., de Vos, R. A., Jansen, E. N., Bratzke, H., and Braak, E. (1998). Neuropathological hallmarks of Alzheimer’s and Parkinson’s diseases. Prog Brain Res 117, 267–5.
Bravo, H. A., and Torres, R. J. (2002). Air pollution levels and trends in the Mexico City metropolitan area. In Urban air pollution and forests: resources at risk in the Mexico City Air Basin (M. Fenn, L. Bauer, and T. Hern´andez, eds.), pp. 121–59. Springer-Verlag, New York.
Brugg, B., Dubreuil, Y. L., Huber, G., Wollman, E. E., Delhaye-Bouchaud, N., and Mariani, J. (1995). Inflammatory processes induce β-amyloid precursor protein changes in mouse brain. Proc Natl Sci USA 92, 3032–5.
Brunekreef, B., and Holgate, S. T. (2002). Air pollution and health. Lancet 360, 1233–42.
Calderon-Garciduenas, L., Azzarelli, B., Acuna, H., Garcia, R., Gambling, T. M., Osnaya, N., Monroy, S., DL Tizapantzi, M. R., Carson, J. L., VillarrealCalder´on, A., and Rewcastle, B. (2002). Air pollution and brain damage. Toxicol Pathol 30, 373–389.
Calderon-Garciduenas, L., Maronpot, R. R., Torres-Jardon, R., Henr´ıquezRold´an, C., Schoonhoven, R., Acu˜na-Ayala, H., Villarreal-Calder´on, A., Nakamura, J., Fernando, R., Reed, W., Azzarelli, B., and Swenberg, J. A. (2003a). DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol Pathol 31, 524–38.
Calderon-Garciduenas, L., Mora-Tiscareno, A., Fordham, L. A., Chung, C. J., Garcia, R., Osnaya, N., Hernandez, J., Acuna, H., Gambling, T. M., Villarreal-Calder´on, A., Carson, J., Koren, H. S., and Devlin, R. B. (2001). Canines as sentinel species for assessing chronic exposures to air pollutants: part 1. Respiratory pathology. Toxicol Sci 61, 342–55.
Calderon-Garciduenas, L., Mora-Tiscareno, A., Fordham, L. A., ValenciaSalazar, G., Chung, C. J., Rodriguez-Alcaraz, A., Paredes, R., Variakojis, D., Villarreal-Calder´on, A., Flores-Camacho, L., Antunez-Solis, A., Henriquez-Roldan, C., and Hazucha, M. J. (2003b). Respiratory damage in children exposed to urban pollution. Pediatr Pulmonol 36, 148– 61.
Cao, C., Matsumura, K., and Watanabe, Y. (1997). Induction of cyclooxygenase- 2 in the brain by cytokines. Ann NY Acad Sci 813, 307–9.
Carson, J. L., Reed, W., Lucier, T., Brighton, L., Gambling, T. M., Huang, C. H., and Collier, A. M. (2002). Axonemal dynein expression in human fetal tracheal epithelium. Amer J Physiol Lung Cell Mol Physiol 282, L421–30.
Dixon, D. A., Tolley, N. D., King, P. H., Nabors, L. B., McIntyre, T. M., Zimmerman, G. A., and Prescott, S. M. (2001). Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest 108, 1657–65.
Dorman, D. C., Brenneman, K. A., McElveen, A. M., Lynch, S. E., Roberts, K. C., and Wong, B. A. (2002). Olfactory transport: a direct route of delivery of inhaled manganese phosphate to the rat brain. J Toxicol Environ Health A 65, 1493–511.
Elmquist, J. K., Breder, C. D., Sherin, J. E., Scammell, T. E., Hickey, W. F., Dewitt, D., and Saper, C. B. (1997). Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol 381, 119– 29.
Finco, T. S., Westwick, J. K., Norris, J. L., Beg, A. A., Der, C. J., and Baldwin, A. S., Jr. (1997). Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem 272, 24113–6.
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., Greenfield, J. P., Haroutunian, V., Buxbaum, J. D., Xu, H., Greengard, P., and Relkin, N. R. (2000). Intraneuronal Aβ42 accumulation in human brain. Am J Pathol 156, 15–20.
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., and Troncoso, J. C. (2001). Intraneuronal Aβ-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125, 489–92.
Hebert, L. E., Scherr, P. A., Bienias, J. L., Bennett, D. A., and Evans, D. A. (2003). Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60, 1119–22.
Hixson, J. E., and Vernier, D. T. (1990). Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31, 545–8.
Ho, L., Purohit, D., Haroutunian, V., Luterman, J. D., Willis, F., Naslund, J., Buxbaum, J. D., Mohs, R. C., Aisen, P. S., and Pasinetti, G. M. (2001). Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch Neurol 58, 487–92.
Junco-Munoz, P., Ottman, R., Lee, J. H., Barton, S. A., Rivas, F., and CerdaFlores, R. M. (1996). Blood lead concentrations and associated factors in residents of Monterrey, Mexico. Arch Med Res 27, 547–51.
Lukiw, W. J., and Bazan, N. G. (1997). Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J Neurosci Res 50, 937–45.
Mattson, M. P., and Camandola, S. (2001). NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107, 247–54.
Moir, R. D., Lynch, T., Bush, A. I., Whyte, S., Henry, A., Portbury, S., Multhaup, G., Small, D. H., Tanzi, R. E., Beyreuther, K., and Masters, C. L. (1998). Relative increase in Alzheimer’s disease of soluble forms of cerebral Aβ amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem 273, 5013–9.
Naslund, J., Haroutunian, V., Mohs, R., Davis, K. L., Davies, P., Greengard, P., and Buxbaum, J. D. (2000). Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 283, 1571–7.
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3, 216– 27.
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., Jones, P. K., Ghanbari, H., Wataya, T., Shimohama, S., Chiba, S., Atwood, C. S., Petersen, R. B., and Smith, M. A. (2001). Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60, 759–67.
Oberdorster, G. (2001). Pulmonary effects of inhaled ultrafine particles. Int Arch Occup Environ Health 74, 1–8.
Oberdorster, G., Sharp, Z., Atudorei, V., Elder, A., Gelein, R., Kreyling, W., and Cox, C. (2004). Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol 16, 437–45.
Osornio-Vargas, A. R., Bonner, J. C., Alfaro-Moreno, E., Martinez, L., GarciaCuellar, C., Ponce-de-Leon-Rosales, S., Miranda, J., and Rosas, I. (2003). Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ Health Perspect 111, 1289–93.
Qin, W., Ho, L., Pompl, P. N., Peng, Y., Zhao, Z., Xiang, Z., Robakis, N. K., Shioi, J., Suh, J., and Pasinetti, G. M. (2003). Cyclooxygenase (COX)-2 and COX-1 potentiate β-amyloid peptide generation through mechanisms that involve γ -secretase activity. J Biol Chem 278, 50970–7.
Rivest, S. (2001). How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology 26, 761–88.
Roth, J., and De Souza, G. E. (2001). Fever induction pathways: evidence from responses to systemic or local cytokine formation. Braz J Med Biol Res 34, 301–14.
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1994). APP gene family: unique age-associated changes in splicing of Alzheimer’s βA4-amyloid protein precursor. Neurobiol Dis 1, 13–24.
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81, 741–66.
Sheng, J. G., Bora, S. H., Xu, G., Borchelt, D. R., Price, D. L., and Koliatsos, V. E. (2003). Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol Dis 14, 133–45.
van Eeden, S. F. and Hogg, J. C. (2002). Systemic inflammatory response induced by particulate matter air pollution: the importance of bone-marrow stimulation. J Toxicol Environ Health A 65, 1597–613.
Xiang, Z., Ho, L., Yemul, S., Zhao, Z., Qing, W., Pompl, P., Kelley, K., Dang, A., Qing, W., Teplow, D., and Pasinetti, G. M. (2002). Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer’s disease neuropathology. Gene Expr 10, 271–8.
Yermakova, A. V., and O’Banion, M. K. (2001). Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer’s disease. Neurobiol Aging 22, 823–36.
Zhou, Y., Su, Y., Li, B., Liu, F., Ryder, J. W., Wu, X., Gonzalez-DeWhitt, P. A., Gelfanova, V., Hale, J. E., May, P. C., Paul, S. M., and Ni, B. (2003). Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Aβ42 by inhibiting Rho. Science 302, 1215–7.