
Acute, subchronic, or chronic exposures to particulate matter (PM) and pollutant gases affect people in urban areas and those exposed to fires, disasters, and wars. Respiratory tract inflammation, production of mediators of inflammation capable of reaching the brain, systemic circulation of PM, and disruption of the nasal respiratory and olfactory barriers are likely in these populations. DNA damage is crucial in aging and in age-associated diseases such as Alzheimer’s disease. We evaluated apurinic/apyrimidinic (AP) sites in nasal and brain genomic DNA, and explored by immunohistochemistry the expression of nuclear factor NFκB p65, inducible nitric oxide synthase (iNOS), cyclo-oxygenase 2 (COX2), metallothionein I and II, apolipoprotein E, amyloid precursor protein (APP), and beta-amyloid1−42 in healthy dogs naturally exposed to urban pollution in Mexico City. Nickel (Ni) and vanadium (V) were measured by inductively coupled plasma mass spectrometry (ICP-MS). Forty mongrel dogs, ages 7 days–10 years were studied (14 controls from Tlaxcala and 26 exposed to urban pollution in South West Metropolitan Mexico City (SWMMC)). Nasal respiratory and olfactory epithelium were found to be early pollutant targets. Olfactory bulb and hippocampal AP sites were significantly higher in exposed than in control age matched animals. Ni and V were present in a gradient from olfactory mucosa > olfactory bulb > frontal cortex. Exposed dogs had (a) nuclear neuronal NFκB p65, (b) endothelial, glial and neuronal iNOS, (c) endothelial and glial COX2, (d) ApoE in neuronal, glial and vascular cells, and (e) APP and β amyloid1−42 in neurons, diffuse plaques (the earliest at age 11 months), and in subarachnoid blood vessels. Increased AP sites and the inflammatory and stress protein brain responses were early and significant in dogs exposed to urban pollution. Oil combustion PM-associated metals Ni and V were detected in the brain. There was an acceleration of Alzheimer’s-type pathology in dogs chronically exposed to air pollutants. Respiratory tract inflammation and deteriorating olfactory and respiratory barriers may play a role in the observed neuropathology. These data suggest that Alzheimer’s disease may be the sequela of air pollutant exposures and the resulting systemic inflammation.
Keywords. Urban pollution; Alzheimer’s disease; ozone; ultrafine particulate matter; brain inflammation; combustion metals; nasal and olfactory pathology; blood–brain barrier; DNA oxidative damage.
INTRODUCTION
Air pollution is a complex mixture of gases, particulate matter (PM), metals, endotoxins, and organic compounds present in outdoor and indoor air. We previously reported that canines exposed to the polluted atmospheric environment in Mexico City (MC) exhibit (a) persistent chronic respiratory tract inflammation, (b) early expression of nuclear neuronal NFκB and endothelial/glial iNOS, (c) breakdown of the nasal and olfactory barriers and the blood–brain barrier (BBB), and (d) early brain microangiopathy (22). Breakdown of the olfactory, respiratory and blood–brain barriers may permit circulating inflammatory mediators, and components of air pollution access to the central nervous system (CNS) (29, 38, 40, 81). Inflammatory processes in the CNS are crucial in neurodegenerative diseases. The presence of inflammatory mediators in the systemic circulation or their upregulation by direct effect upon target brain areas (ie, olfactory bulb) may lead to an activation of endothelial, glial, and microglial cells, which in turn could increase the formation of reactive oxygen species (ROS) and upregulate “protective” and “harmful” genes (39). Significant levels of DNA damage arise from endogenous and exogenous cellular sources, mostly from intermediates of oxygen reduction that either attack the bases or the deoxyribosyl backbone of DNA. These oxidized bases are predominantly repaired by a base excision repair pathway (30). During base excision repair, DNA glycosylase cleaves a modified DNA base at the glycosyl bond resulting in the formation of an apurinic/apyrimidinic (AP) site. AP sites inhibit DNA replication, give rise to base substitution mutations, and loss of genetic integrity (68). Nakamura et al recently established a method to quantitate the number of AP sites using an Aldehyde Reactive Probe (ARP) (78), which can react with the aldehydic forms of AP sites caused by DNA glycosylase or spontaneous depurination/depyrimidination, resulting in the formation of stable biotin-tagged complexes. The identification of aldehydic AP sites using ARP enabled us to quantitate the total number of regular AP sites by the ARP-slot blot (ASB) assay (78). Although ARP is not a specific probe for AP sites, as it can also react with aldehydic bases and probably aldehydic oxidized deoxyribose chemical alterations, it is a good indicator of DNA oxidative damage. Oxidative damage and the presence of significant numbers of AP sites in target anatomical areas contributes to mutations, chromosome aberrations and transcription errors (68), and likely may play a role in neurodegenerative disorders.
Based on our previous results (22) where nuclear NFκB activation was followed by strong expression of endothelial, glial, and neuronal iNOS in urban pollution-exposed dogs, we hypothesized that AP sites would be increased in tissues such as nasal mucosa and olfactory bulb in exposed animals, and that there would be concomitant robust expression of inflammatory markers such as COX2, along with increased immunoreactivity of Apo E and APP, and deposition of β-amyloid1−42 in areas such as frontal cortex and hippocampus. Further, since these dogs were exposed to PM with a significant amount of metals derived from combustion of fossil fuels (75), and since the literature suggests that after nasal instillation of a metal-containing solution, transport of the metal via olfactory axons can occur rapidly (38, 46, 103), we hypothesized that combustion-related metals such as Ni and V would be present in nasal and brain tissues.
Our results suggest that dogs naturally exposed to urban air pollutants in Mexico City exhibit significant DNA damage in the first synaptic relay from the olfactory epithelium, and in the hippocampus, and combustion-associated metals such as Ni and V are detected in nasal and brain areas. There is an early activation of transcription factor-kappa B, and induction of a range of genes encoding injury-responsive proteins including iNOS, APP, ApoE, β amyloid, COX2, and metallothionein I-II. Our major premise is that neurodegenerative diseases such as Alzheimer’s disease could begin early in life with air pollutants playing a crucial role.
MATERIALS AND METHODS
Canine Population
Fourteen controls and 26 MC healthy mongrel dogs (15 M/25 F) were studied (Table 1). The younger MC dogs (≤1 year) were whelped and housed in an outdoor-indoor kennel, husbandry was in compliance with American Association of Laboratory Animal Certification Standards. Dogs kept in an outdoor-indoor kennel were housed 1 dog per run, fed once a day (Purina Dog Chow, Ralston-Purina, Cuautitlan, Mexico), and had water ad libitum. They were under daily veterinary observation during their entire life, and at no time was there any evidence of overt respiratory, cardiovascular, or neurological diseases. The remaining older MC dogs were from within 5 miles of the air pollution monitoring station, and all the exposed and control dogs were home-raised and continuously kept outdoors. These home-raised dogs were periodically seen by a veterinarian, had all the applicable vaccines (including distemper), and were dewormed regularly. They were fed twice a day with a combination of dog chow and home food leftovers and had free access to potable water. These dogs had no contact with other animals, they lived permanently in the selected residential area and had no exposures to local toxics sources (paints, metals, solvents, etc). Their health histories were unremarkable throughout their lives. None of the dogs had been outside their residential area or lived in a different city.

The selection of the control and exposed cities was based on the similar altitude above sea level (22), and our previous clinical and pathology data from canines and children from the selected cities (19–21). The rationale behind selecting the southwest geographical area in Mexico City lies in 2 major factors: (a) the spacial distribution of pollutants such PM10, O3, SO2, and NO2 within the southwest residential area is a result of wind transport of the mass precursor pollutants emitted in the industrial north and central parts of Mexico City, and (b) evidence of nasal and pulmonary pathology in healthy children and adults as well as cytokine imbalance in children living in SWMMC (19–21). Tlaxcala was selected because its altitude above sea level is similar to Mexico City and studies in dogs from these area have shown minimal pathology in lungs and hearts (19, 20). The study protocol was reviewed and approved by the Institutional Basic Research Committee at the Instituto Nacional de Pediatria in Mexico City. Euthanasia was conducted in accordance with established guidelines (87).
Study Areas
Polluted City: MC is a megacity with approximately 20 million inhabitants, 3.5 million vehicles, more than 30,000 industrial facilities, 12,000 commercial/service consuming fuel facilities and an undetermined number of dry cleaning, printing, and solvent facilities. Daily emissions of fixed and mobile sources, combined with the basin climatologic and topographic factors contribute to the polluted atmosphere. Ozone is the most important air pollutant in terms of frequency, occurrence, high levels, and spatial distribution coverage. An average of 4 ± 1 hours/day of O3 above 0.08 ppm is recorded in SWMMC year-round. Both PM10 and PM2.5 exceed their respective annual arithmetic means above the standards (annual US National Ambient Air Quality Standards (NAAQS) PM10 50 µg/m3, and PM2.5 15 µg/m3): 78 µg/m3 and 21.6 µg/m3 (33). Quarterly average concentrations of lead in total suspended particles (TSP) have shown a downtrend pattern from a range of 0.85 to 2.6 µg/m3 in 1990 to less than 0.4 µg/m3 since 1995 (Pb USNAAQS standard quarterly average: 1.5 µg/m3). Other pollutants detected in SWMMC include: volatile organic compounds, formaldehyde and acetaldehyde; mutagenic PM, alkane hydrocarbons, various metals eg, nickel, vanadium, manganese, chromium, and peroxyacetyl nitrate (5, 33, 75, 111). In addition, lipopolysaccharides (LPS), and endotoxins are detected in PM10 (12). For the purpose of this work, Mexico City’s atmospheric pollutant data was obtained from a representative pollutant monitoring station located in Southwest MC, and the available literature.
Control City: Tlaxcala is a small 41-km2 city located at 2,230 meters above sea level and 130 km East of MC. The city is separated from the meteorological and climatic pattern of the Mexico valley influenced by the eastern portion of the same chain of mountains that forms this valley. As a consequence of its location, the ventilation of the region is sustained and continuous. Tlaxcala has 68,000 inhabitants and 260 small industrial facilities, most of them not requiring combustion processes. Due to the combination of the relatively few contributing emission sources from industry and cars, and the good ventilation conditions by the regional wind, air pollution levels in Tlaxcala are below the current US standards.
Necropsy and Tissue Preparation
Animals were examined by a veterinarian before they were euthanized. A gross external description was done after their demise with special focus on age changes and nutritional status, followed by complete necropsies. Immediately after death, the skull was opened and the olfactory bulbs and the brain removed. The trachea, extrapulmonary bronchi and lungs, along with the heart were excised intact from the thoracic cavity, and then sampled. The abdominal cavity was opened and examined; samples of liver, spleen, and kidneys were obtained. The nasal respiratory and olfactory mucosa, olfactory bulbs, selected brain areas from alternating right and left cerebral hemispheres, and selected extra CNS tissues were immediately frozen in liquid nitrogen and kept at −80◦C.
Sections adjacent to the frozen material were immersed in 10% neutral formaldehyde, fixed for 48 hours, and transferred to 70% alcohol. Tissues were consistently sampled from all dogs. Sections were taken from olfactory bulbs, entorhinal cortex, superior frontal gyrus, superior parietal gyrus, superior temporal gyrus, hippocampus, head of the caudate nucleus, dorsal medial nucleus of thalamus, and hypothalamus at the same level as the previous section, neocerebellum, midbrain at the level of the superior colliculus, the red nucleus and the medial longitudinal fasciculus, pons, and medulla. Frozen tissue for the AP sites and ICP-MS were taken from the cortex and the white matter taking care of making a perpendicular cut to the brain surface and keeping similar amounts of cortex and white matter for each method. Paraffin sections 5-µm thick were cut and routinely stained with hematoxylin and eosin (H&E). The terminal deoxynucleotidyl transferase (TdT) labeling assay (TUNEL, ApopTag, Intergen Company, New York, NY) was used to assess cells with DNA strand breaks (64). Immunohistochemistry (ICH) was performed using previously described protocols (22) with proliferating cell nuclear antigen (PCNA) clone PC10, Dako, Carpinteria, CA ; β-amyloid precursor protein (APP), β-amyloid (1–42), and apolipoprotein E (APOE), BD Transduction Laboratories; NFκB p65 and anti-macNOS (macrophage inducible iNOS) Santa Cruz Biotechnology; glial fibrillary acidic protein (GFAP) Boehringer Mannheim, and cyclo-oxygenase 2 human polyclonal antibody Cayman Chemical, MI, USA. The brain histopathologic parameters evaluated included: the presence of histological elements characteristic of neuronal and glial cell death, the distribution and characteristics of astrocytes (on H&E, GFAP, and NFκB p65), and vascular changes (H&E). The ICH observations and the histopathologic severity of the brain findings was assessed semiquantitatively using the 0, +, ++, +++ scoring system. The evaluation of the olfactory epithelium was done using the terminology suggested by Hardisty (44) for rat olfactory tissues. Sections were read blindly by experienced observers with no access to the codes regarding the geographical source of the animals.
DNA Extraction and Aldehyde Slot Blot (ASB) Assay
DNA extraction and the ASB assay were done for the 14 control and the 26 MC exposed dogs by procedures reported by Nakamura (78, 79). AP sites are expressed per 1,000,000 nucleotides (79).
Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
Tissue from olfactory epithelium, olfactory bulbs, frontal cortex, and/or peribronchial lymph nodes was taken from control (n = 3) and MC (n = 4) dogs for ICP-MS. Each sample was weighed and digested using a closed vessel microwave digestion in the presence of nitric acid and H2O2. The resulting solutions were analyzed directly for their Ni and V content. Several quality control samples, including reagent blanks were also analyzed. The recovery of added Ni and V in the low-level, and high-level method controls were 73%/114%, and 105%/99.5%, respectively. Ni and V concentrations were reported in µg/g of wet tissue.
Statistics
Statistics were performed using the Stata Program (College Station, Texas). Differences in the numbers of AP sites between control and exposed dogs (combined sexes) were tested with the 2-Sample Wilcoxon rank-sum test. A regression model using the AP site differences between age-matched dogs was designed to test the differences in hippocampus AP sites in age-matched control vs. exposed dogs. Significance was assumed at p < 0.05. Data are expressed as mean values ± SD.
RESULTS
Air Quality Data
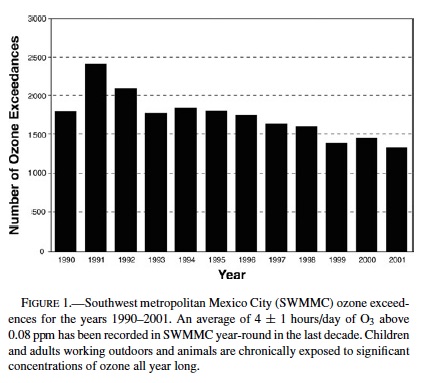
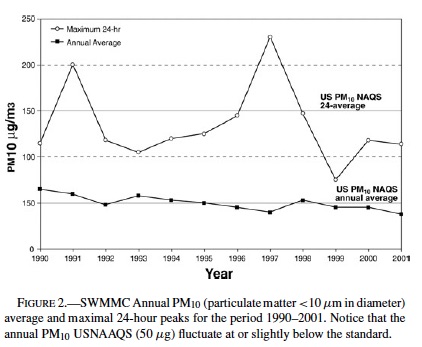
Figure 1 shows the annual number of hourly O3 exceedences in SWMMC during the period 1990–2001. The SWMMC atmosphere is characterized by average maximal ozone daily concentrations of 0.250 ppm. An average of 4 ± 1 hours/day with ozone >0.08 ppm is recorded in SWMMC year-round (83.9% of days). Figure 2 shows the maximum 24-hour PM10 and the annual average concentrations registered during the period 1990–2001 in SWMMC. Both PM10 and PM2.5 annual concentrations fluctuate at or slightly below the standard.
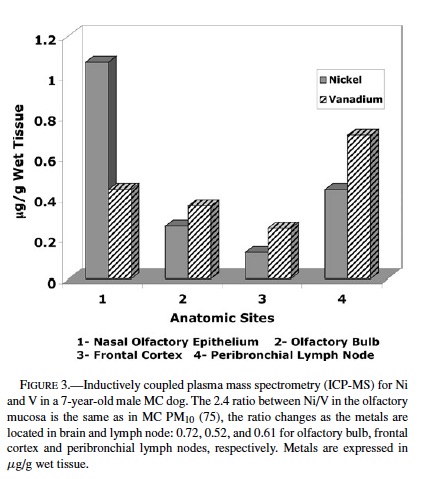
ICP-MS. Nickel and vanadium were detected in each anatomical region examined in 3 controls and 4 exposed animals (Table 2). On average vanadium concentrations in exposed animals were higher than in control dogs (controls 0.19 ± 0.17 vs. exposed 0.36 ± 0.3), although the difference was not statistically significant. Vanadium concentrations were higher than Ni in all frontal cortices in exposed animals, so that the ratio Ni/V was less than 1. The Ni/V ratio in the frontal cortex of control dogs was greater than 1 in 2/3 dogs. In 1 representative exposed dog where tissues from olfactory epithelium, olfactory bulb, and frontal cortex were available for ICP-MS, there was a gradient in the concentrations of both Ni and V going from higher concentrations in the olfactory epithelium to lower, but still detectable levels in the frontal cortex (Figure 3). The average concentrations of both metals in the peribronchial lymph nodes were higher than in olfactory bulb or frontal cortex in both controls and exposed animals.

AP Sites in Nasal Respiratory and Olfactory Epithelium, Brain, Lungs, and Heart
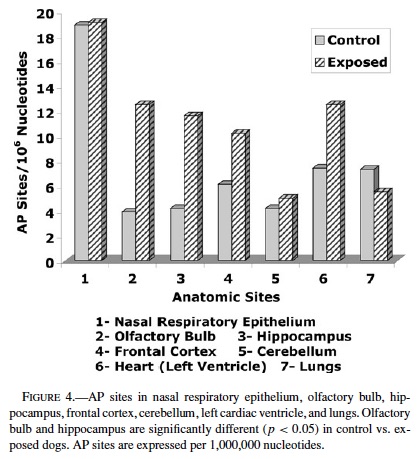
AP sites were significantly higher in MC dogs compared with controls in the olfactory bulb (controls 3.9 ± 0.8 vs. MC 12.5 ± 1.7 p = 0.0002) (Figure 4). AP sites in hippocampus increased with age in both controls and MC dogs, however the increment was significant in MC dogs vs. controls closely age-matched after age 1 y (p < 0.002). No significant differences in AP sites in age-matched dogs were seen in nasal respiratory mucosa (18.9 ± 4.4 vs. 19.1 ± 2.8), frontal cortex (6.6 ± 1 vs. 10.2 ± 3.2), cerebellum (4.2 ± 0.5 vs. 5.0 ± 0.6), left cardiac ventricle (7.4 ± 0.7 vs. 12.5 ± 3), and lungs (7.3 ± 0.7 vs. 5.5 ± 0.6).
Clinical and Gross Pathological Observations
Animals in this study were all healthy, and there were no overweight or undernourished dogs.
Non-CNS Relevant Findings: Lungs from SWMMC dogs had bronchiolar epithelial and smooth muscle hyperplasia. Chronic mononuclear cell infiltrates along with macrophages filled with PM surrounded the bronchiolar walls and extended into adjacent vascular structures. Peribronchial lymph nodes were grossly pigmented and filled with PM. Dogs from Tlaxcala had mild bronchiolar epithelial hyperplasia, small numbers of alveolar macrophages with PM, and rare foci of inflammatory cells in association with either terminal bronchioles or pulmonary blood vessels. Peribronchial lymph nodes showed small clusters of PM-containing macrophages.
Nasal Histopathology
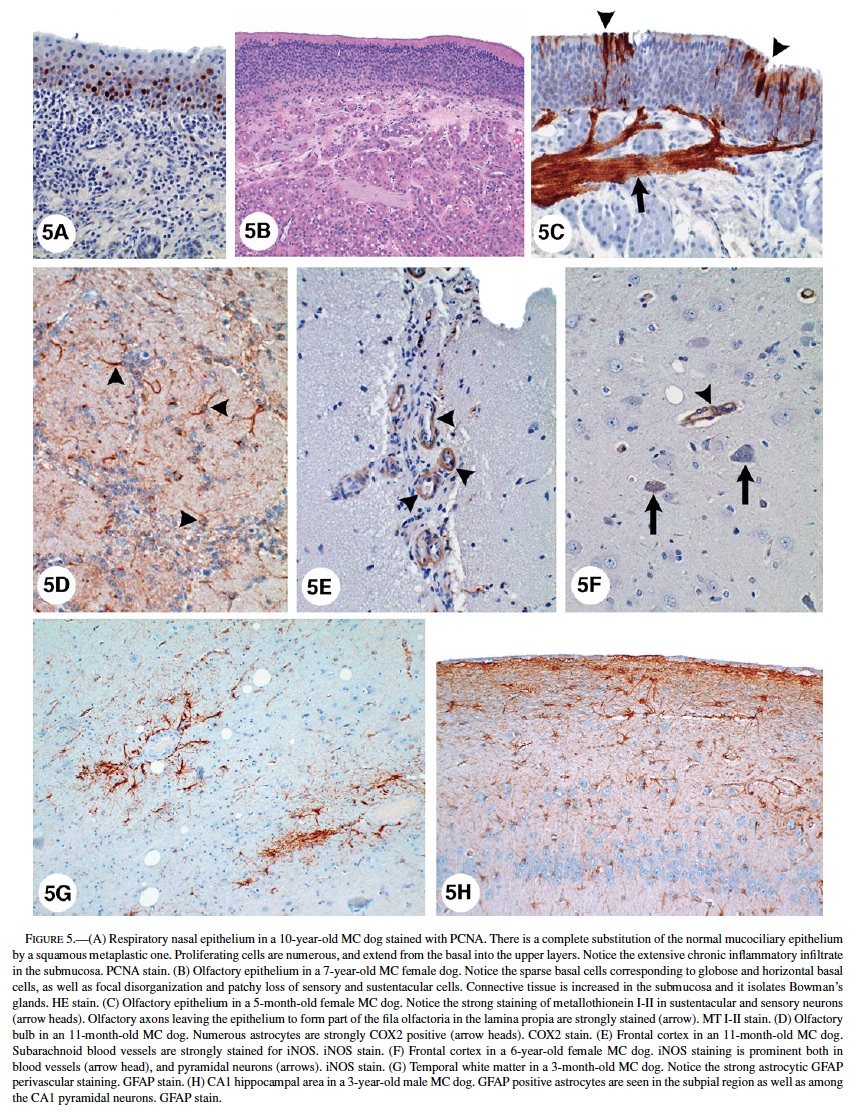
Respiratory Epithelium: Control dogs (>5 years) showed unremarkable pseudostratified ciliated columnar epithelium with goblet cells, while older dogs had focal areas of squamous metaplasia and mild to moderate thickening of the basement membrane (BM). SWMMC dogs younger than 3 months had unremarkable respiratory nasal epithelium. Changes in SWMMC dogs were progressively worse with age and were characterized by extensive replacement of the mucociliary epithelium by squamous metaplasia. Increased proliferation (PCNA positivity) of metaplastic squamous cells was prominent (Figure 5A). Although no dysplastic changes were seen, regenerative metaplastic squamous cells with abundant hyalinized cytoplasm and large nuclei were seen in some dogs. There was an increased number of small blood vessels and tufts of basement membrane-like material projecting into the squamous metaplastic mucosa. In a few cases, submucosal glands were dilated and atrophic, and there was marked thickening of the basement membrane along with a chronic, mononuclear inflammatory infiltrate in the submucosa.
Olfactory Epithelium: A summary of the histological findings according to the Hardisty classification (44) is seen in Table 3. The neurosensory epithelium was intact and well defined in all control dogs and in MC dogs younger than 3 months. The sharp boundaries between respiratory and olfactory epithelia present in control animals and in young MC dogs became irregular and disrupted with age in exposed dogs. Microscopic changes were detected in SWMMC dogs, with the earliest seen in 8-month-old dogs. The changes were multifocal and characterized by abnormal orientation of the sensory olfactory and sustentacular cells, as well as degeneration with loss of cells, resulting in a decrease in the thickness of the neuroepithelium (Figure 5B). Older SWMMC dogs show patchy areas with severe to complete loss of basal, sustentacular, and sensory cells. PCNA-positive cells in the basal layer were rare in the most affected areas. Metallothionein I-II immunopositivity was strong in neuronal, and sustentacular cells (Figure 5C). Olfactory axons forming part of the fila olfactoria in the lamina propia were strongly stained for metallothionein (Figure 5C). Tubuloacinar olfactory glands were intact in younger SWMMC and control animals; however, MC dogs older than 1 year showed scattered hyperchromatic nuclei and deposition of particulate material in the glandular cytoplasm. Areas with decreased numbers of atrophic Bowman’s glands surrounded by proliferating connective tissue were focally prominent (Figure 5B). Metallothionein I-II weak, patchy immunopositivity was seen in control dogs older than 5 y.

Brain Pathology
CNS Gross Findings: All dogs were mesaticephalic. The remaining cerebral hemisphere was cut on the coronal plane and there was no evidence of ventricular dilatation or cortical atrophy in any animal.
Brain Histopathology: Intergroup differences (ie, control versus Mexico City dogs) in immunohistochemical staining are reported in Table 4. The following represents a generalized description of the patterns of staining.

Olfactory Bulbs: TUNEL-positive glial cells were rare in control animals and few in exposed animals in the different layers of the olfactory bulbs. Exposed dogs exhibited reactive astrocytosis (GFAP-positive) in all olfactory bulb layers (including external and internal plexiform, mitral cell layers, and the olfactory glomeruli). GFAP varied in intensity (+ to +++) but was seen in every SWMMC dog starting at age 8 months. Animals older than 4 years had marked increases in GFAP, particularly in the glomerular region. PCNA showed isolated proliferating cells in the external and internal plexiform layers. Astrocytes in the external and internal plexiform layers and at the junction with the mitral cell layer had strong staining (+++) with metallothionein I-II. Olfactory nerve bundles were also strongly stained. COX 2 stained the cytoplasm of mitral and granular neurons and astrocytes (Figure 5D). Scattered iNOS immunopositive endothelial cells were seen in exposed dogs. Except for a few reactive astrocytes in the olfactory glomeruli of older control dogs, the olfactory bulbs were unremarkable.
In MC dogs nuclear immunoreactivity to the p65 subunit of NF-κB was present in cortical neurons, dentate gyrus, Purkinje, cerebellar granular, and cortical perivascular cells. The NF-κB protein complex was localized in the cytosol of reactive astrocytes in MC dogs ≥1 year, as indicated by the morphological similarity of NFκB-positive cells and reactive GFAP-positive astrocytes. Nuclear NFκB immunoreactivity was not seen in control dogs. MC dogs also had strong iNOS staining in endothelial, white matter, and subpial astrocytes; frontal, parietal, and temporal cortical neurons in layers III–V; and the lateral choroid plexus epithelium (Figures 5E, 5F). Strong nuclear staining was also seen in hypothalamus, dorsal medial nucleus of thalamus, midbrain, substancia nigra pars compacta, cranial nerve III, VI, VII, and VIII nuclei, and pons reticular formation. Control dogs older than 6 years showed weak iNOS positivity in endothelial and white matter glial cells in the cortex.
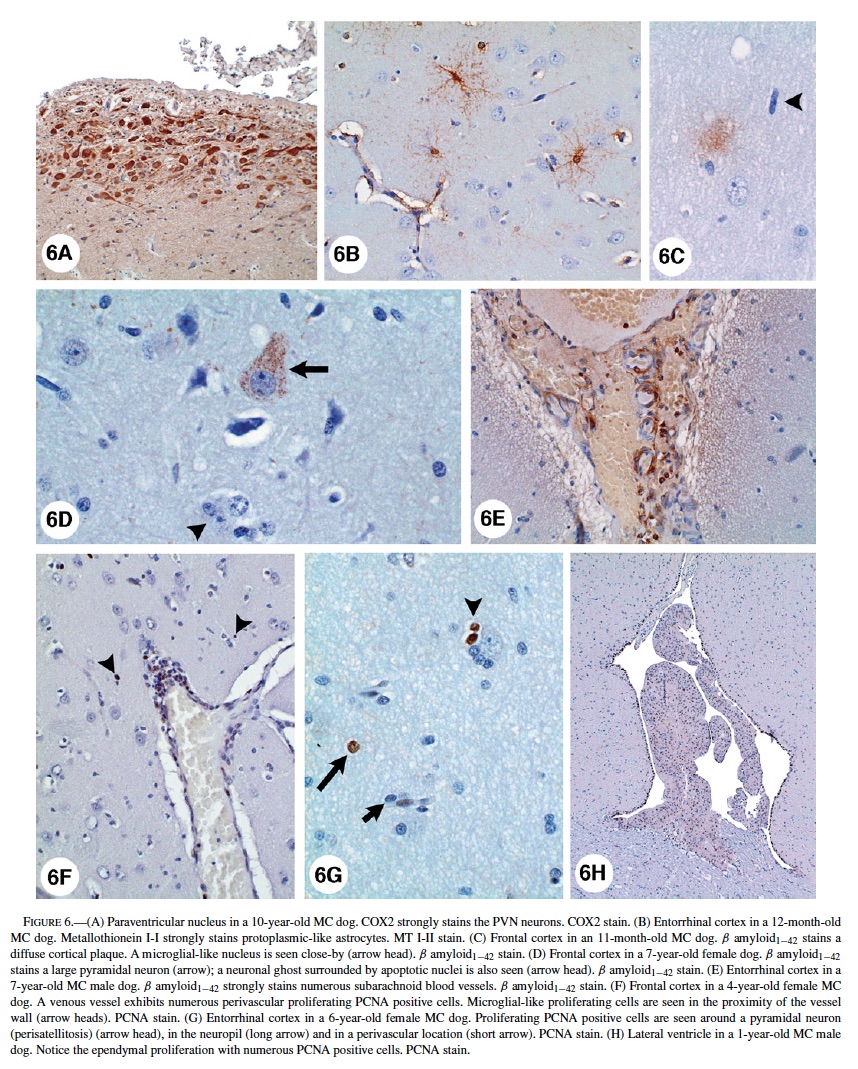
Astrocytes with a small amount of eosinophilic cytoplasm were seen around blood vessels and neurons in young SWMMC dogs. These astrocytes were predominantly cortical and did not stain with GFAP. Reactive GFAP-positive astrocytes were focally prominent in subpial areas and perivascular deep white matter of all SWMMC animals starting at age 3 months (Figure 5G). Scattered discrete foci of GFAP-positive astrocytes were seen in the gray matter and increased with age. This observation was particularly prominent in hippocampus CA1 (Figure 5H). Foci of neuronal glial satellitosis were observed in the cortical gray matter, and were more prominent in frontal and entorhinal cortices. COX2 strong neuronal staining was present in frontal, temporal and parietal MC dogs’ cortices. In the hippocampi, strongly positive COX2 neurons included the CA1-2 and CA4 regions, and dentate gyri; while in the hypothalamus, the paraventricular nucleus was strongly stained (Figure 6A). COX2 positive astrocytic and endothelial cells could also be seen in the white matter, and to lesser extent in the gray and in the subpial region. COX2 immunopositivity was present in Bergmann glia, brainstem large motor cranial nerve neurons, and substantia nigrae.
Control dogs had a similar distribution of COX2, however, the staining was minimal, and confined to neurons. Entorhinal cortex protoplasmic astrocytes in exposed dogs had strong immunoreactivity with metallothionein I-II (Figure 6B). ApoE immunopositivity was seen in cortical neurons (predominantly nuclear staining), astrocytes (gray and white matter), perivascular macrophages, and endothelial cells and pericytes in small arterioles in the white matter. APP was present in neuronal and glial cytoplasm, smooth muscle cells in subarachnoid arteries, as well diffusely deposited in the frontal and temporal cortices, hippocampus, subiculum and presubiculum neuropil. Rare positive β-amyloid1−42, and APP nonneuritic plaques were seen in frontal and entorhinal cortices in young exposed dogs starting at age 11 months (Figure 6C). β-amyloid1−42 selectively accumulated in the perikaryon of pyramidal frontal neurons as discrete granules (Figure 6D), and was present in endothelial capillary cortical cells, subarachnoid and cortical blood vessels, and microglia-like cells (Figure 6E). Microglial-like cells with ameboid profiles were present in the cortex and the white matter (Figure 6C).
Ameboid microglia could be seen as early as 8 months in SWMMC dogs, however, their number increased with age particularly in the gray matter, including the subiculum, presubiculum and basal ganglia. Clear-cut strongly PCNA positive cells were seen in the neuropil, as well in perivascular locations, suggesting migration of cells undergoing proliferation from the blood vessels to the brain parenchyma (Figure 6F). Neural progenitor cell clusters in the subventricular areas in the lateral ventricles showed PCNA-positive cells in MC dogs as old as 8 years. Both in the gray and white matter of MC dogs an increased number of S-phase glial cells were present (Figure 6G). A 1-year-old male dog from MC showed 2 foci of cellular proliferation in the supratentorial brain, involving hyperplastic ependyma in the lateral ventricle (Figure 6H), and undifferentiated cells in the lateral ventricle choroid plexus. TUNEL-positive glial nuclei in close relationship with neurons, and in perivascular areas were present in frontal, temporal, and parietal cortices of SWMMC dogs. No cortical neurons showed positive TUNEL nuclei, however a few were positive for PCNA suggestive of DNA damage and /or repair. TUNEL-positive cells were seen in the olfactory bulb in both controls and exposed dogs. Control animals exhibited a rare TUNEL-positive nucleus in the cortical white matter.
DISCUSSION
Clinically healthy canines exposed to urban air pollutants, particularly ozone and PM, show chronic inflammation in the upper and lower respiratory tract, significant damage to the nasal respiratory and olfactory epithelium, and higher AP sites in the olfactory bulb (the first synaptic relay in the olfactory pathway) and hippocampus than age matched animals residing in an urban environment in compliance with US NAAQS standards. An early robust IHC expression of stress and inflammation response proteins, astrogliosis, and deposition of β-amyloid1−42 in diffuse plaques were prominent findings in the brain of young <1-year-old MC dogs. Vanadium and nickel, present in the soluble and exchangeable fraction of PM, were detected by ICP-MS indicating that brain uptake of metals associated with fossil fuel combustion may occur in a natural exposure setting. Metal uptake could be through olfactory neurons and axons, peripheral sensory nerves, direct passage of inhaled particles into systemic blood circulation, and through lung intravascular macrophage-like cells that ingest ultrafine PM and are capable of reaching the brain (20, 34, 38, 44, 46, 81).
In the aging dog, brain lesions sharing morphological characteristics with Alzheimer’s disease develop spontaneously, and β-amyloid deposition in dogs older than 10 years correlates with cognitive decline (1, 4, 13, 28, 108). Old dogs show neurofibrillary tangles, and vascular, perivascular and neuronal hydroxynonenal protein, as well as a decrease in glutamine synthetase and glutathione activity indicative of oxidative damage (45, 89). The neuropathological changes we described in SWMMC dogs have the morphological characteristics of the Alzheimer-like changes in old dogs; however, we have documented diffuse β-amyloid plaque development in very young animals along with significant DNA damage in target sites as evidenced by the number of AP sites. The relevance of increased brain DNA damage in the context of exposure to air pollutants is crucial in view of the role of oxidative stress as a potential causal factor in aging and neurodegenerative diseases (10, 17, 41, 49, 89, 95, 106).
Oxidative stress is important in the Alzheimer’s disease (AD) brain (84), and it is postulated that β-amyloid leads to neuronal lipid peroxidation, protein and DNA oxidation (17, 48, 97). Increased AP sites in the olfactory bulb were higher in MC pups as young as 4 months, while in the hippocampus major increases started at 4 years of age. Thus, significant DNA damage is present early in the olfactory bulb in the absence of β-amyloid1−42, several months before β-amyloid cortical diffuse plaques, and several years before positive β-amyloid subarachnoid blood vessels are detected. In MC dogs the olfactory mucosa histopathology appeared by age 4 months, and thus may serve as a window for the olfactory bulb pathology. This is an important piece of information considering that in neurodegenerative diseases such as Alzheimer’s and Parkinson’s olfaction is impaired in the earlier stages (47, 59, 65, 107). Interestingly, AP sites were not significantly different in controls versus exposed in areas such as frontal cortex, an observation we explain based on the young age of the majority of dogs studied. On the other hand, AP sites in cerebellum, an anatomical region mostly spared from AD hallmarks, showed no difference between controls and exposed animals.
Our working hypothesis illustrated in Figure 7 proposes that air pollution could produce brain damage through several potential pathways including: induction of upper respiratory, lung epithelial, and endothelial injury leading to persistent chronic inflammation. The endothelial and epithelial injury are accompanied by the production of cytokines such as IL6, IL1β, IL10, TNFα, and interferon γ , which in turn affect the expression of adhesion molecules and chemokines (IL8) (55, 110). We have clinical evidence of such chronic inflammation in the respiratory tract, and systemic involvement in SWMMC children. Seemingly healthy children ages 5–17 years exhibit important chest x-ray alterations, lung function, and peripheral blood changes, and an imbalanced cytokine network (21). Pro- and anti-inflammatory cytokines can be transported across the blood brain barrier, can activate or disrupt the BBB, and trigger cascades leading to the activation of MAP kinases/NFκB or the JAK/STAT transduction pathways in vascular-associated CNS cells (8, 40, 43, 74, 88, 90, 91).
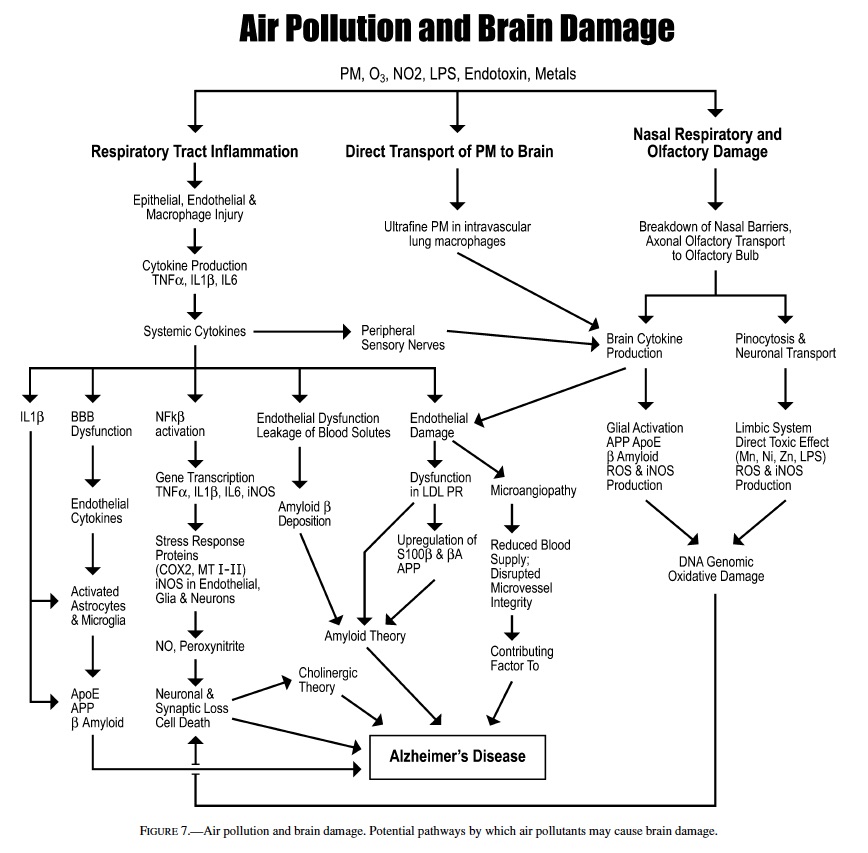
Brain–blood vessels exhibit constitutive and induced expression of receptors for TNFα, IL1β, and IL6 (35, 77). Thus, the bloodstream cytokines such as TNFα and IL1β have the ability to trigger NFκB-inducible cytokines within the blood– brain barrier (91). Elmquist (34) suggests an alternate pathway: cytokines could signal the CNS via sensory nerves such as the vagus. An intriguing possibility in air pollution exposed subjects, since both LPS (present in PM and likely related to the 500 tons of canine fecal material deposited everyday in MC streets) and IL1β (increased in the serum of MC clinically healthy children, unpublished observations, CalderonGarciduenas 2003) are recognized by chemosensory receptors located in vagal paraganglia in the vagus nerve at several levels including cervical, thoracic, and abdominal regions (34). Thus, in pollution-exposed subjects CNS cytokine-triggering could take place via signaling through the vagus nerve.
The transcription factor NFκB is activated in MC dogs as young as 2 weeks (22). NFκB is an inducible regulator of a broad range of genes, plays a pivotal role in brain cell death and survival pathways (9, 83), and has a crucial role in regulating cytokine cascades including TNFα and IL6, as well as β APP (72). Heavy metals such as vanadium and nickel can activate NFκB in the brain (26). Vanadium crosses the blood– brain barrier (67), and it can produce direct oxidative DNA damage by catalyzing ROS formation and indirect damage by inflammatory cells recruited by vanadium-induced expression of leukocyte chemoattractants (56). Nickel suppresses ROS scavenging mediated by glutathione and protein-bound sulfhydryl groups, resulting in increased concentrations of ROS, enhanced lipid peroxidation, DNA damage, and altered calcium homeostasis (101).
High-level activation of NFκB may promote the sustained production of neurotoxins, and indeed activation of NFκB in astrocytes results in increased expression of nitric oxide synthase and nitric oxide production (51). Early iNOS endothelial expression is likely of primary importance in the chain of events’ pathogenesis, by contributing to the maintenance, self-perpetuation and progression of the neurodegenerative processes (41). Nitric oxide (NO) opens the BBB leading to vasogenic edema and secondary brain damage (24, 106). Strong expression of iNOS in endothelial cells and astrocytes, namely 2 major components of the blood–brain barrier—the defensive front of the brain—are crucial findings in pollution-exposed dogs. Progressive subpial, and white and grey matter astrogliosis were prominent early features in exposed animals raising the issue of the role of glial-neuronal interactions in the neuropathology of these canines. Griffin (42) put forward the hypothesis that chronic activation of glial inflammatory processes arising from environmental insults to neurons, starts a cytokine cycle of cellular and molecular events with neurodegenerative consequences. We think that such a cytokine cycle could play a role in the neuropathological findings observed in these canines with lifetime exposures to air pollutants. Neurons interact with glial cells and rely on substances released by glia for their normal functions (80, 100). Glial pathology affects cortical neurons and also the subcortical cholinergic neurons that project to the cerebral and hippocampal cortices to maintain cortical neuronal activity (80). Chronic glial reactivity in the model of fetal cortical tissue grafted heterotopically onto the superior colliculus is accompanied by an increase in apoE and APP levels (71). Reactive astrocytes are a primary source of APP in these grafts (25), and chronic gliosis is associated with altered processing of the amyloid precursor protein in vivo and could initiate pathological changes associated with AD (6). Canines in SWMMC exhibited white matter perivascular gliosis as early as 3 months of age, and the astrogliosis increased with age to a greater extent than in age-matched controls from Tlaxcala.
An interesting aspect of this study in exposed animals is the early and robust expression of critical intermediates of the acute phase response to infection and inflammation such as COX2, and anticytotoxic systems such as metallothionein I-II. MC dogs showed intense COX2 immunostaining in olfactory bulb astrocytes, cortical endothelial, neuronal and glial cells, and in the paraventricular nucleus of the hypothalamus that controls the neuroendocrine system (91). COX2 isoform is undetectable in most tissues under basal conditions but intense transcriptional activation can be observed in macrophages by endotoxin LPS and proinflammatory cytokines (63), and in brain capillaries in response to intravenous TNFα and IL1β (60). COX2 is found exclusively in neurons in the normal, unstimulated brain (15), and is rapidly regulated in developing and adult rat forebrain by physiological synaptic activity (15, 54). COX2 has been assigned detrimental effects based on proinflammatory actions, as well as cardioprotective effects in a model of ischemia/reperfusion injury (11). In the rat model of transient middle cerebral artery occlusion, neuronal upregulation of COX2 contributes to ischemic damage (82), and leptomeningeal exposure to LPS increases expression of COX2 protein in the meninges causing COX2- dependent dilatation of cerebral arterioles in vivo (16).
The robust expression of COX2 in MC dogs is important since as a primary inflammatory mediator that converts arachidonic acid into precursors of vasoactive prostaglandins; and production of ROS is an important component of the conversion process (7, 102). Early and sustained production of COX2 may play a role in free-radical mediated cellular damage, alter cellular metabolism, and produce vascular dysfunction; these are all crucial in neurodegenerative diseases, such as Alzheimer’s disease (3, 86). Metallothioneins on the other hand, are small, cysteine-rich heavy metal-binding proteins (70) involved in the protection of tissues against various forms of oxidative injury. Zinc and copper, glucocorticoids, catecholamines, endotoxin, and cytokines are involved in the induction of brain MT I-II (32). MC animals displayed strong MT I-II immunostaining in the olfactory epithelium, olfactory axons leaving the epithelium to form part of the fila olfactoria, olfactory bulb, and protoplasmic astrocytes in entorrhinal cortex, and hippocampus. In healthy dogs raised in open air, MTI-II immunoreactivity is observed in olfactory epithelium sustentacular cells, and in astrocytes of all layers of the olfactory bulb (98). MTI-II is also seen in hypertrophic astrocytes associated with a variety of brain lesions in dogs, suggesting that MT is also involved in repair of injured neural tissues (99).
In normal human developing and aged brains, MTs are located in astrocytes (104, 105). In AD patients there is an increase in MT immunoreactivity in the olfactory epithelium (27). The overexpression of MTs in a damaged olfactory epithelium and olfactory bulb in MC dogs is most likely a protective mechanism, and, as an acute phase protein, could be related to circulating cytokines such as IL-1 (57), and the presence of endotoxins in urban air that come in direct contact with the olfactory epithelium. Heavy metals also induce metallothionine expression. The olfactory bulb is well known to accumulate certain metals with increased avidity (103) and can transport metals to cortical areas (46). Ni and V were present in the olfactory epithelium, olfactory bulb, and frontal cortex of MC dogs, but were also detected in control dogs that did not overexpress MTI-II. Thus, exposure to these metals alone did not evoke MT-II overexpression. It remains to be determined whether Ni and V accumulation interacts with other pollutants in the upregulation of MTI-II expression. The detection of Ni and V in control dogs from a low polluted area may be due to exposure through the soil or other unidentified sources of both soluble and insoluble metals (ie, volcanic ash) (46).
In keeping with the respiratory tract inflammation and associated inflammatory cytokines, APP is detected in young MC dogs. APP levels are influenced by IL1 and IL6 (66, 92, 94) and are thought to be important contributors to the pathogenesis of Alzheimer’s disease (69, 73). APP is crucial in neuronal stress responses in vivo, and mediates the kinesin-I dependent axonal transport of cargo proteins such as presenilin-1 (52, 76). ApoE on the other hand, limits the Aβ-induced glial neuroinflammatory response (61), exerts a neuroprotective effect against ischemia through its antioxidant action (58), and it has been identified in canines in relation with early cerebrovascular amyloid deposits (109). Finally, accumulation of β-amyloid intraneuronally (114) and in diffuse plaques were very early findings in exposed dogs.
A potentially important observation in exposed dogs was the increased number of PCNA-positive S-phase glial cells both in the white matter and to lesser extent in the gray matter compared to age matched control dogs. Several mechanisms could account for the increase in PCNA-positive glial cells in exposed dogs: (a) the modulation of glial cell proliferation by cytokines (93), (b) axonal degeneration causing a transient up-regulation of glial cell proliferation and GFAP protein expression (97), (c) carcinogenic metals such as vanadium and nickel activating NFκB (26), and (d) NFκB targeting genes that regulate cell proliferation, apoptosis and cell migration (53). These observations, in addition to the incidental findings of ependymal hyperplasia and an undifferentiated choroid plexus tumor in a highly exposed SWMMC dog, raise the issue of the role of environmental pollutants and neural carcinogenesis. A recent epidemiologic report from Mexico City, showing an increased number of SWMMC boys with central nervous system tumors mostly of astrocytic origin (36), mandates further study this potential association.
Ozone and particulate matter (PM) concentrations in Mexico City are above the US current standards, and an unrelenting haze blankets the city. Heavy metals in the atmosphere are present in aerosol form (fine PM fraction) and include fuel oil-associated metals: Ni, V, Cr, and S, as well as Zn, Cu, and Mn, elements associated to industry or traffic. Organic dusts, lipopolysaccharides, and endotoxins in PM10 are a large pollutant contributor, mostly coming from 500 tons of dog feces deposited daily on the streets (23). Regulation and assessment of pollution have been focused on ambient concentrations; however, indoor pollution is also significant and represent an important contribution to overall PM exposure (50, 112, 113).
The nasal respiratory and olfactory findings in this work emphasize the potential impact of upper respiratory tract physiology and the metabolic xenobiotic detoxification (29, 62) for inhaled gases and materials in the nose when the barriers are not longer intact. MC children and canines show indirect evidence of attempts to restore and protect the nasal epithelium against the effects of continuous exposure to inhaled pollutants (18). The observation of ultrafine PM deposition in nasal epithelial cells and intercellular spaces, and in the transudate between epithelial cells in nasal biopsies from healthy children in MC (18), their greater inflammatory potential when deposited in the lung (85), and the fact that ultrafine PM can be transported in the systemic circulation via macrophage-like cells (19), highlights the importance of ultrafine particles in the study of neurodegenerative diseases. More importantly, the respiratory tract chronic inflammatory process elicited upon exposure to air pollutants (outdoor and indoor) including fine and ultrafine PM (2, 18, 20, 31, 37, 81, 85) could serve as the trigger for a chain of events that involves a cytokine circuit cascade in the brain. Dr. R. Floyd refers to a “spark” to repeatedly set off the inflammatory brain cascades (39), the sustained exposure to air pollutants could be the “spark” needed to damage the brain.
We fully realize that the issue of exposure to pollutants is crucial to support our findings and to arrive at conclusions that can be extrapolated to human populations. Within the context of this study, selection of dogs was based on their known residency and clinical history. Exposed dogs were all from SWMMC and remained outdoors within a 5-mile radius of the pollution monitoring station where exposure to urban pollution was unavoidable. We selected a small control city, Tlaxcala, which lacks any industrial infrastructure, is in compliance with US NAAQS standards, and is at the same altitude as Mexico City. We have a significant amount of data from previous studies in dogs and in children in Tlaxcala that document an absence of pollution-related pathology (18–22). Thus, we feel that the issue of “treatment” is adequately defined for this study.
In summary, we described canines exposed to urban concentrations of ozone and PM and a myriad of other air pollutants that exhibit early brain genomic DNA damage in target areas, brain deposition of metals related to combustion processes, persistent activation of NFκB, strong expression of iNOS, early neuronal and β-amyloid plaque accumulation, and robust inflammatory and stress protein brain responses. These canine findings are of sufficient magnitude and clinical significance to warrant concern that similar pathology may be occurring at an accelerated pace in humans residing in large metropolitan areas or those exposed to significant amounts of PM such as the result of wildfires, disasters, or war events. Neurodegenerative disorders such as Alzheimer’s disease may relate to air pollutant exposures.
ACKNOWLEDGMENTS
This work was supported in part by NIEHS grants T-32 ES07017, P30-ES10126, and P-30-CA 16086. Special thanks to Raquel Garcia, Silvia Monroy, Lina Romero, Norma Osnaya, and Maria del Rosario Tizapantzi for their support.
REFERENCES
1. Adams B, Chan A, Callahan H, Milgram NW (2000). The canine as a model of human cognitive aging: Recent developments. Prog NeuroPsychopharmacol Biol Psychiat 24: 675–692.
2. Adamson IY, Vincent R, Bjarnason SG (1999). Cell injury and interstitial inflammation in rat lung after inhalation of ozone and urban particles. Am J Respir Cell Mol Biol 20: 1067–1072.
3. Aisen PS (2002). Evaluation of selective COX2 inhibitors for the treatment of Alzheimer’s Disease. J Pain Symp Manag 23: 835–840.
4. Anderson AJ, Ruehl WW, Fleichmann LK, Stenstrom K, Entriken TL, Cummings BJ (2000). DNA damage and apoptosis in the aged canine brain: relationship to Aβ deposition in the absence of neuritic pathology. Prog Neuro-Psychopharmacol Biol Psychiat 24: 787–799.
5. Baez AP, Belmont R, Padilla H (1995). Measurements of formaldehyde and acetaldehyde in the atmosphere of Mexico City. Environ Pollut 89: 163–167.
6. Bates KA, Fonte J, Robertson A, Martins RN, Harvey AR (2002). Chronic gliosis triggers Alzheimer’s disease-like processing of amyloid precursor protein. Neuroscience 113: 785–796.
7. Bazan NG, Colangelo V, Lukiw WJ (2002). Prostaglandins and other lipid mediators in Alzheimer’s disease. Prost Lip Med 68–69: 197–210.
8. Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P (2000). Interleukin-1beta-induced changes in blood brain barrier permeability, apparent difussion coeficcient, and cerebral blood volume in rat brain: A magnetic resonance study. J Neurosci 20: 8153–8159.
9. Blondeau N, Widmann C, Lazdunski M, Heurteaux C (2001). Activation of the nuclear factor-kappa B is a key event in brain tolerance. J Neurosci 21: 4668–4677.
10. Boje KM, Arora PK (1992). Microglial-produced nitric oxide and reactive oxides mediate neuronal cell death. Brain Res 587: 250–256.
11. Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, Dawn B (2002). Discovery of a new function of cyclooxygenase (COX)-2: COX2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res 55: 506–516.
12. Bonner JC, Rice AB, Lindroos PM, O’Brien PO, Dreher KL, Rosas I, Alfonso-Moreno E, Osornio-Vargas AR (1998). Induction of the lung myofibroblast PDGF receptor system by urban ambient particles from Mexico City. Am J Respir Cell Mol Biol 19: 672–680.
13. Borras D, Ferrer I, Pumarola M (1999). Age-related changes in the brain of the dog. Vet Pathol 36: 202–211.
14. Borroni B, Akkawi N, Martini G, Colciaghi F, Prometti P, Rozzini L, Di Luca M, Lenzi GL, Romanelli G, Caimi L, Padovani A (2002). Microvascular damage and platelet abnormalities in early Alzheimer disease. J Neurol Sci 203–204: 189–193.
15. Breder CD, Dewitt D, Kraig RP (1995). Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol 355: 296–315.
16. Brian JE, Moore SA, Faraci FM (1998). Expression and vascular effects of cyclooxygenase-2 in brain. Stroke 29: 2600–2606.
17. Butterfield DA, Drake J, Pocernich C, Castegna A (2001). Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β peptide. Trends Mol Med 7: 548–553.
18. Calderon-Garciduenas L, Valencia-Salazar G, Rodrıguez-Alcaraz A, Gambling TM, Garcıa R, Osnaya N, Villarreal-Calderon A, Devlin RB, ´ Carson JL (2001). Ultrastructural nasal pathology in children chronically and sequentially exposed to air pollutants. Am J Respir Cell Mol Biol 24: 132–138.
19. Calderon-Garciduenas L, Gambling TM, Acuna H, Garcia R, Osnaya N, Monroy S, Villarreal-Calderon A, Carson J, Koren HS, Devlin RB (2001). Canines as sentinel species for assessing chronic exposures to air pollutants: Part 2. Cardiac pathology. Toxicol Sci 61: 356–367.
20. Calderon-Garciduenas L, Mora-Tiscareno A, Fordham LA, Chung CJ, Garcia R, Osnaya N, Hernandez J, Acuna H, Gambling TM, Villarreal- Calderon A, Carson J, Koren HS, Devlin RB (2001). Canines as sentinel species for assessing chronic exposures to air pollutants: Part 1. Respiratory pathology. Toxicol Sci 61: 342–355.
21. Calderon-Garciduenas L, Mora-Tiscareno A, Fordham LA, Valencia G, Chung CJ, Rodriguez-Alcaraz A, Paredes R, Variakojis D, VillarrealCalderon A, Flores-Camacho L, Antunez-Solis A, Henrıquez-Roldan C, Hazucha MJ (2003). Respiratory pollution damage in children. Pediatric Pulmonol 36: 148–161.
22. Calderon-Garciduenas L, Azzarelli B, Acuna H, Garcia R, Gambling TM, Osnaya N, Monroy S, Tizapantzi R, Carson JL, Villarreal-Calderon A, Rewcastle B (2002). Air pollution and brain damage. Toxicol Pathol 30: 373–389.
23. Cano D (2001). Heces caninas grave problema de salud. El Universal, Mexico City, May 17: 4.
24. Chao CC, Hu S, Moliter TW, Shaskan EG, Peterson PK (1992). Activated microglia mediate cell injury via a nitric oxide mechanism. J Immunol 149: 2736–2741.
25. Chauvet N, Apert C, Dumoulin A, Epelbaum J, Alonso G (1997). Mab32C11 antibody to amyloid precursor protein recognizes a protein associated with specific astroglial cells of the rat central nervous system characterized by their capacity to support axonal outgrowth. J Comp Neurol 377: 550–564.
26. Chen F, Shi X (2002). Intracellular signal transduction of cells in response to carcinogenic metals. Crit Rev Oncol Hematol 42: 105–121.
27. Chuah MI, Getchell ML (1999). Metallothionein in olfactory mucosa of Alzheimer’s disease patients and apoE deficient mice. Neuroreport 10: 1919–1924.
28. Cummings BJ, Head E, Ruehl W, Milgram NW, Cotman CW (1996). Canine as an animal model of human aging and dementia. Neurobiol Aging 17: 259–268.
29. Dahl AR, Lewis JL (1993). Respiratory tract uptake of inhalants and metabolism of xenobiotics. Annu Rev Pharmacol Toxicol 33: 383–407.
30. Demple B, Harrison L (1994). Repair of oxidative damage to DNA: Enzymology and biology. Annu Rev Biochem 63: 915–948.
31. Driscoll KE, Carter JM, Hassenbein DG, Howard B (1997). Cytokines and particle-induced inflammatory cell recruitment. Environ Health Perspect 105: 1159–1164.
32. Ebadi M, Iversen PL, Hao R, Cerutis DR, Rojas P, Happe HK, Murrin LC, Pfeiffer RF (1995). Expression and regulation of brain metallothionein. Neurochem 27: 1–22.
33. Edgerton SA, Bian X, Doran JC, Fast JD, Hubbe JM, Malone EL, Shaw WJ, Whiteman CD, Zhong S, Arriaga JL, Ortiz E, Ruiz M, Sosa G, Vega E, Limon T, Guzman F (1999). Particulate air pollution in Mexico City:A Collaborative Research Project. Air Waste Manag Assoc 49: 1221– 1229.
34. Elmquist JK, Breder CD, Sherin JE, Scamell TE, Hickey WF, Dewitt D, Saper CB(1997). Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunorectivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol 381: 119–129.
35. Ericsson A, Liu C, Hart RP, Sawchenko PE (1995) Type-1 interleukin-1 receptor in the rat brain: Distribution, regulation, and relationship to sites of IL1-induced cellular activation. J Comp Neurol 361: 681–698.
36. Fajardo-Gutierrez A, Navarrete-Martinez A, Reynoso-Garcia M, ZarzosaMorales ME, Mejia-Arangure M, Yamamoto-Kimura LT (1997). Incidence of malignant neoplasms in children attending social security hospitals in Mexico City. Med Pediatr Oncol 29: 208–212.
37. Ferin J, Oberdorster G, Penney DP (1992). Pulmonary retention of ultra ¨ fine and fine particles in rats. Am Respir Cell Mol Biol 6: 535–542.
38. Feron VJ, Arts JH, Kuper CF, Slootweg PJ, Woutersen RA (2001). Health risks associated with inhaled nasal toxicants.Crit Rev Toxicol 31: 313–347.
39. Floyd RA (1999). Neuroinflammatory processes are important in neurodegenerative diseases: An hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Rad Biol Med 26: 1346–1355.
40. Gloor SM, Wachtel M, Bolliger MF, Ishihara H, Landmann R, Frei K (2001). Molecular and cellular permeability control at the blood-brain barrier. Brain Res Rev 36: 258–264.
41. Grammas P, Botchlet TR, Moore P, Weigel PH (1997). Production of neurotoxic factors by brain endothelium in Alzheimer’s disease. Ann NY Acad Sci 826: 47–55.
42. Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI (1998). Glial-neuronal interactions in Alzheimer’s disease: The potential of a cytokine cycle in disease progression. Brain Pathol 8: 65–72.
43. Griffin WS, Mrak RE (2002). Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol 72: 233–238.
44. Hardisty JF, Garman RH, Harkema JR, Lomax LG, Morgan KT (1999). Histopathology of nasal olfactory mucosa from selected inhalation toxicity studies conducted with volatile chemicals. Toxicol Path 27: 618–627.
45. Head E, Liu J, Hagen TM, Muggenburg BA, Milgram NW, Ames BN, Cotman CW (2002). Oxidative damage increases with age in a canine model of human brain aging. J Neurochem 82: 375–381.
46. Henriksson J, Tallkvist J, Tjalve H (1997). Uptake of nickel into the brain ¨ via olfactory neurons in rats. Toxicol Lett 91: 153–162.
47. Hirai T, Kojima S, Shimada A, Umemura T, Sakai M, Itakura C (1996). Age-related changes in the olfactory system of dogs. Neuropath Appl Neurobiol 22: 531–539.
48. Hung MT, Yamada K, Nabeshima T (2002). Amyloid β-peptide induces cholinergic dysfunction and cognitive deficits: A minireview. Peptides 23: 1271–1283.
49. Iwase K, Miyanaka K, Shimizu A, Nagasaki A, Gotoh T, Mori M, Takiguchi M (2000). Induction of endothelial nitric-oxide synthase in rat brain astrocytes by systemic lipopolysaccharide treatment. J Biol Chem 275: 11929–11933.
50. Johnson T, Long T, Ollison W (2000). Prediction of hourly microenvironmental concentrations of fine particles based on measurements obtained from the Baltimore scripted activity study. J Expo Anal Environ Epidemiol 10: 403–411.
51. Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA (1993). Constitutive NFkB in neurological disorders. Mol Aspects Med 14: 171–190.
52. Kamal A, Almenar-Queralt A, LeBlanc JF, Roberst EA, Goldstein LSB (2001). Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and persenilin-1 requires APP. Nature 414: 643– 648.
53. Karin M, Cao Y, Greten FR, Li ZW (2002). NFϕB in cancer: From innocent bystander to major culprit. Nat Rev Cancer 2: 301–310.
54. Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P (1996). COX- 2 a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci USA 93: 2317– 2321.
55. Kawanami O, Jiang HX, Mochimaru H, Yoneyama H, Kudoh S, Ohkuni H, Oami H, Ferrans VJ (1995). Alveolar fibrosis and capillary alteration in experimental pulmonary silicosis in rats. Am J Respir Crit Care Med 151: 1946–1955.
56. Kawanishi S, Hiraku Y, Murata M, Oikawa S (2002). The role of metals in site-specific DNA damage with reference to Carcinogenesis. Free Rad Biol Med 32: 822–832.
57. Kikuchi Y, Irie M, Kasahara T, Sawada JI, Terao T (1993). Induction of metallothionein in a human astrocytoma cell line by interleukin-1 and heavy metals. FEBS 317: 22–26.
58. Kitagawa K, Matsumoto M, Kuwabara K, Takasawa KI, Tanka S, Sasaki T, Matsushita K, Ohtsuki T, Yanagihara T, Hori M (2002). Protective effect of Apolipoprotein E against ischemic neuronal injury is mediated through antioxidant action. J Neurosci Res 68: 226–232.
59. Kovacs T, Cairns NJ, Lantos PL (2001). Olfactory centres in Alzheimer’s disease: Olfactory bulb is involved in early Braak’s stages. Neuroreport 12: 285–288.
60. Lacroix S, Rivest S (1998). Effect of acute systemic inflammatory response and cytokines on the transcription of the genes encoding cyclooxygenase enzymes (COX1 and COX2) in the rat brain. J Neuroch 70: 452–466.
61. LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, Guo L, Van Eldik LJ (2001). Apolipoprotein E and apolipoprotein E receptors modulate A β-induced glial neuroinflammatory responses. Neurochem Int 39: 427– 434.
62. Larsson P, Tjalve H (2000). Intranasal instillation of a ¨ flatoxin B1 in rats: Bioactivation in the nasal mucosa and neuronal transport to the olfactory bulb. Toxicol Sci 55: 383–391.
63. Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H, Liou S, Simmons D, Hwang D (1992). Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysacchararide. J Biol Chem 267: 25934–25938.
64. Levin S, Bucci TJ, Cohen SM, Fix AS, Hardisty JF, LeGrand EK, Maronpot RR, Trump BF (1999). The nomenclature of cell death: Recommendations of an ad hoc Committee of the Society of Toxicologic Pathologists. Toxicol Pathol 27: 484–490.
65. Liberini P, Parola S, Spano PF, Antonini L (2000). Olfaction in Parkinson’s disease: Methods of assessment and clinical relevance. J Neurology 247: 88–96.
66. Licastro F, Campell L, Kincaid C, Veibbergs I, Uden EV, Rockenstein E, Mallory M, Gilbert JR, Masaliah E (2000). Apolipoprotein E and alpha-1-antichymotrypsin allele polymorphism in sporadic and familiar Alzheimer’s disease: Peripheral inflammation or signals from the brain? J Neuroimmunol 103: 97–102.
67. Li S, Zhang T, Yang Z, Gou X (1991). Distribution of vanadium in tissues of nonpregnant and pregnant Wistar rats. J West China 22: 196–200.
68. Loeb LA, Preston BD (1986). Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet 20: 201–230.
69. Lucca U, Tettamanti M, Forline G, Spagnoli A(1994). Nonesteroidal antiinflammatory drugs in Alzheimer’s disease. Biol Psychiatry 36: 854–856.
70. Margoshes M, Vallee BL (1957). A cadmium protein from equine kidney cortex. J Am Chem Soc 79: 4813–4814.
71. Martins RN, Taddel K, Kendall C, Evin G, Bates KA, Harvey AR (2001). Altered expression of apolipoprotein E, amyloid precursor protein and presenilin-1 is associated with chronic reactive gliosis in rat cortical tissue. Neuroscience 106: 557–569.
72. Mattson MP, Camandola S (2001). NFκB in neuronal plasticity and neurodegenerative disorders. J Clin Inv 107: 247–254.
73. McGeer PL, McGeer EG (1995). The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev 21: 195–218.
74. Mrak RE, Griffin ST (2001). Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Ageing 22: 903–908.
75. Mugica V, Maubert M, Torres M, Munoz J, Rica E (2002). Temporal and spatial variations of metal content in TSP and PM10 in Mexico City during 1996–1998. J Aerosol Sci 33: 91–102.
76. Muller U, Kins S (2002). APP on the move. ¨ Trends Mol Med 8: 152–155.
77. Nadeau S, Rivest S (1999). Effects of circulating tumor necrosis factor (TNF) on the neuronal activity and expression of the genes encoding the TNF receptors (p55 and p75) in the rat brain: A view from the blood– brain-barrier. Neuroscience 93: 1449–1464.
78. Nakamura J, Walker VE, Upton PB, Chaing SY, Kow YW, Swenberg JA (1998). Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res 58: 222–225.
79. Nakamura J, Swenberg JA (1999). Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res 59: 2522–2526.
80. Nelson PG, McCune SK, Ades AM, Nelson KB (2001). Glial-neurotrophic mechanisms in Down syndrome. J Neural Trans Suppl 61: 85–94.
81. Nemmar A, Hoet PHM,Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B (2002). Passage of inhaled particles into the blood circulation in humans. Circulation 105: 411–414.
82. Nogawa S, Zhang F, Ross E, Iadecola C (1997). Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neuroscien 17: 2746–2755.
83. Nomura Y (2001). NF-κB activation and IϕBα dynamism involved in iNOS and chemokine induction in astroglial cells. Life Sci 68: 1695– 1701.
84. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001). Oxidative damage is the earliest event in Alzheimer’s. J Neuropath Exp Neurol 60: 759–767.
85. Oberdorster G (2001). Pulmonary effects of inhaled ultra ¨ fine particles. Int Arch Occup Environ Health 74: 1–8.
86. Oka A, Takashima S (1997). Induction of cyclooxygenase 2 in brains of patients with Down’s syndrome and dementia of Alzheimer’s type: Specific localization in affected neurons and axons. Neuroreport 8: 1161–1164.
87. Panel on Euthanasia (1993). J Am Vet Med Assoc 202: 229–249.
88. Pan W, Kastin AJ (2001). Upregulation of the transport system for TNF alpha at the blood brain barrier. Arch Physiol Biochem 109: 350–353.
89. Papaioannou N, Tooten PC, van Ederen AM, Bohl JR, Rofina J, Tsangaris T, Gruys E (2001). Immunohistochemical investigation of the brain of aged dogs: I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein an oxidative damage product, en senile plaques. Amyloid 8: 11–21.
90. Rivest S, Lacroix S, Vallieres L, Nadeau S, Zhang J, Laflamme N (2000). How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammation and infectious stimulae. Proc Soc Exp Biol Med 223: 22–38.
91. Rivest S (2001). How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinol 26: 761–788.
92. Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LNG (1999). Translation of the Alzheimer amyloid precursor protein mRNA is upregulated by interleukin-1 through 5-untranslated region sequences. J Biol Chem 274: 6421–6431.
93. Ruhl A, Farnzke S, Stremmel W (2001). IL1 beta and IL10 have dual ¨ effects on enteric glial cell proliferation. Neurogast Mot 13: 89–94.
94. Salbaum JM, Weidmann A, Masters CL, Beyreuther K (1988). The promoter of Alzheimer’s disease amyloid A4 precursor gene. EMBO J 7: 2807–2813.
95. Sayre IM, Smith MA, Perry G (2001). Chemistry and biochemistry of oxidative stress in neurodegenerative diseases. Curr Med Chem 8: 721– 738.
96. Schwob JE, Youngentob SL, Ring G, Iwema CL, Mezza RC (1999). Reinnervation of the rat olfactory bulb after methyl bromide-induced lesion: Timing and extent of reinnervation. J Comp Neurol 412: 439–457.
97. Selkoe DJ (2001). Alzheimer’s disease: Genes, proteins and therapy. Physiol Rev 81: 741–766.
98. Shimada A, Irie M, Kojima S, Kobayashi K, Yamano Y, Umemura T (1996). Immunohistochemical localization of metallothionein in the olfactory pathway of dogs. J Vet Med Sci 58: 983–988.
99. Shimada A, Uemura T, Yamamura Y, Kojima S, Morita T, Umemura T (1998). Localization of metallothionein I and II in hypertrophic astrocytes in brain lesions of dogs. J Vet Med Sci 60: 351–358.
100. Schubert P, Ogata T, Marchini C, Ferroni S (2001). Glia-related pathomechanisms in Alzheimer’s disease: A therapeutic target? Mech Ageing Develop 123: 47–57.
101. Stohs SJ, Bacchi D (1995). Oxidative mechanisms in the toxicity of metal ions. Free Rad Biol Med 18: 321–336.
102. Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK (2000). Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma 17: 695–711.
103. Sunderman FW (2001). Nasal toxicity, carcinogenicity, and olfactory uptake of metals. Annals Clin Lab Sci 31: 3–24.
104. Susuki K, Nakajima K, Otaki N, Kimura M (1994). Metallothionein in developing human brain. Biol Sig 3: 188–192.
105. Susuki K, Nakajima K, Otaki N, Kimura M, Kawaharada U, Uchara K, Hara F, Nakazato Y, Takatama M (1994). Localization of metallothionein in aged human brain. Pathol Int 44: 20–26.
106. Thiel VE, Audus KL (2001). Nitric oxide and blood–brain barrier integrity. Antioxid Redox Signal 3: 273–278.
107. Tissingh G, Berendse HW, Bergmans P, De Waard R, Drukarch B, Stoof JC, Wolters EC (2001). Loss of olfaction in de novo and treated Parkinson’s disease: Possible implications for early diagnosis. Mov Dis 16: 41–46.
108. Torp R, Head E, Cotman CW (2000). Ultrastructural analyses of β-amyloid in the aged dog brain: Neuronal β-amyloid is localized to the plasma membrane. Prog Neuro-Psychopharmacol Biol Psychiat 24: 801– 810.
109. Uchida K, Kuroki K, Yoshino T, Yamaguchi R, Tateyama S (1997). Immunohistochemical study of constituents other than beta-protein in canine senile plaques and cerebral amyloid angiopathy. Acta Neuropathol 93: 277–284.
110. Vaillant P, Menard O, Vignaud JM, Martinet N, Martinet Y (1996). The role of cytokines in human lung fibrosis. Monaldi Arch Chest Dis 51: 145–152.
111. Villalobos-Pietrini R, Blanco S, Gomez-Arroyo S (1995). Mutagenicity assessment of airborne particles in Mexico City. Atmospheric Environ 89: 163–167.
112. Wainman T, Zhang J, Weschler CJ, Lioy PJ (2000). Ozone and limonene in indoor air: A source of submicron particle exposure. Environ Health Perspect 108: 1139–1145.
113. Williams R, Suggs J, Zweidinger R, Evans G, Creason J, Kwok R, Rodes C, Lawles P, Sheldon L (2000). The 1998 Baltimore Particulate Matter Epidemiology-Exposure Study: Part 1. Comparison of ambient, residential outdoor, indoor and apartment particulate matter monitoring. J Expo Anal Environ Epidemiol 10: 518–532.
114. Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001). Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Letters 306: 116– 120.