A short-term 5-day nose-only cigarette smoke exposure study was conducted in Fisher 344 rats to identify smoke-induced tracheal protein changes. Groups of 10 male and female 5 week old rats were assigned to 1 of 4 exposure groups. Animals received filtered air, or 75, 200 or 400 mg total particulate matter (TPM)/m3 of diluted 3R4F Kentucky reference cigarette mainstream smoke. Exposures were conducted for 3 hrs/day, for 5 consecutive days. Tracheas from half the rats were processed for pathology, and tracheas from the other half of the rats frozen immediately for proteomics. We hypothesized that smoke will activate tracheal inflammatory, apoptotic, proliferative, and stress-induced pathways. Mucosal epithelial toxicity from the inhaled material was evidenced by cilia shortening and loss of tracheal mucosal epithelium in smoke-exposed animals. Mucosal thinning occurred in all smoke-exposed groups with hyperplastic reparative responses in the 200 and 400 mg TPM/m3 groups. Tracheal lysates from control vs. treated animals were screened for 800 proteins using antibody-based microarray technology and subsequently the most changed proteins evaluated by Western blot. Tracheal proteins expressed at high levels that were markedly increased or decreased by smoke exposure depended on dose and gender and included caspase 5, ERK 1/2 and p38. Signaling pathways common between the morphologic and protein changes were stress, apoptosis, cell cycle control, cell proliferation and survival. Changes in identified proteins affected by smoke exposure were associated with tracheal mucosal pathology, may induce functional tracheal changes, and could serve as early indicators of tracheal damage and associated disease.
Keywords:
cigarette smoke, proteomics, trachea, cell signaling, apoptosis
Introduction
Trachea has been used as a model of in vitro toxicity studies, for neoplastic transformation and for regulatory testing1,2,3. Rat tracheal epithelial cells are a common model but in vitro assays lack the 3-D features of cellular interactions in invivo conditions. The tracheal epithelium, analyzed as a model for use in clinical testing, was more sensitive to smoking, responding with threefold more differentially expressed genes than small airway epithelium4. The trachea transcriptome paralleled the small airway epithelium, with 156 of 167 (93%) genes significantly up- and downregulated by smoking in the small airway epithelium showing similar direction and magnitude of response to smoking. Thus, trachea epithelium may also serve as a surrogate clinical model for smoke-induced changes in the small airway epithelium. One advantage of this model is that trachea epithelium can be obtained without extensive sedation, representing a less invasive surrogate “canary” for smoking-induced changes4.
Long-term animal studies for assessment of toxicity and carcinogenesis are not ideal models because of the requirement for long-term intensive smoke exposure, the length of time required to induce visible tumors, and the lack of an established robust animal model for cigarette smoke-mediated carcinogenesis. We are evaluating a newer mechanistic approach that focuses on induction of toxicological changes relevant to disease such as cell proliferation, chronic inflammation, and inhibition of apoptosis5, 6. Toxicoproteomics is being employed to evaluate the protein changes associated with these functional and phenotypic changes.
The purpose of the present study was to evaluate changes in trachea pathology and proteins in nose-only exposed male and female Fischer 344 rats exposed to mainstream whole cigarette smoke. We hypothesized that the proteins that were most affected by short-term smoke exposure would fit into pathways involved in inflammation, proliferation, stress responses, and changes in cell morphology. In this study, we present the profiles of differentially expressed proteins in trachea tissue from smoke-exposed vs controls and relate the changed biological processes to mechanisms of disease development. Assessment of toxicological changes and candidate biomarkers in short-term animal studies could provide a new approach for evaluation of the mechanism(s) of toxin action as well as new assays for product testing and disease diagnoses.
Materials and Methods
Animal exposure
The study protocol was approved by the Institutional Animal Care and Use Committee at the Illinois Institute of Technology Research Inst. (IITRI). Forty male and 40 female Fischer 344 rats at 5 weeks of age were divided into groups of 10 male and 10 female rats and exposed to either filtered air (Air Control) or 75, 200 or 400 mg total particulate matter (TPM)/m3 of diluted cigarette smoke generated from 3R4F Kentucky Reference Research Cigarettes. Exposures were for 3 hrs/day, for 5 consecutive days. Scheduled necropsies were conducted on study day 5 immediately after final exposure.
Tissue processing/histopathological evaluations
Tracheas were collected from all animals. Tracheas from half the animals were infused with and immersed in neutral buffered formalin for 24 hrs and then placed in 70% ethanol and processed for pathology. Trachea tissues were embedded in paraffin, 5 μm sections were obtained, and tissues were processed by routine histological methods, stained with hematoxylin and eosin, and evaluated microscopically by a board-certified veterinary pathologist. The remaining 5 tracheas from the other half of the animals were frozen immediately in liquid nitrogen for proteomic analysis. The whole trachea tissue samples were lysed in 250 μl of Lysis Buffer with Triton X-100 by sonication. The supernatants were recovered for Bradford assays before being stored at –80°C. All the above procedures were carried out on ice. The whole trachea lysates from 5 pooled animals per group were evaluated as described below using Microarray and Western blotting analyses.
Proteomics evaluation: antibody-based protein microarray
The Kinex Antibody Microarray KAM 1.0 analyses were performed with detergent-solubilized protein lysates7. Briefly, protein lysates from control and treated rat trachea tissues were labeled with a fluorescent dye and unincorporated dye removed by ultrafiltration. Purified labeled proteins from the control and its correspondingly treated sample were incubated simultaneously on opposite sides of a Kinex KAM-1.0 antibody microarray (Kinexus Bioinformatics, Vancouver, B.C. Canada). Each Kinex antibody microarray is based on duplicate measurements with 2 identical fields of antibody grids containing 800 signaling proteins. Arrays were scanned using a ScanArray scanner with a resolution of 10 μm, and the resulting images were quantified using ImaGene 8.0 (BioDiscovery, El Segundo, CA). The values for the duplicate measurements were averaged. Proteins that were selected as valid to move to the next screening step fit into at least one of the following categories: (1) Proteins showed large changes in >1 microarray, (2) Proteins were altered in >1 treatment group, (3) Proteins that had highest % change from control values and (4) Proteins that were previously found in high quantity in rat tissue by searching the KiNET database (http://www.kinexus.ca/) which contains large amounts of information about protein changes in various species of animals. The KiNET DataBank is an open-access, on-line DataBank with antibody-based measurements of hundreds of signaling proteins and phosphosites in over 10,000 different tissues and specimens.
Determination of protein changes induced by smoke
The top 108 antibodies that demonstrated smoke-induced changes and fit at least 1 of the 4 criteria stated above moved to a prescreen status for pre-validation. The pooled samples were evaluated to assess which target proteins were detectable by immunoblotting in trachea lysates. This step and the next step of Western blotting were necessary, because the proteins evaluated in the KinexTM KAM 1.0 antibody microarrays were not denatured, which increases the opportunity for false positives and false negatives due to antibody cross-reactivity, protein-protein interactions, and blocked epitopes in protein complexes.
Western blotting was performed with pooled samples from 5 tracheas per treatment group to determine the presence of the 108 target proteins using methods detailed previously7. Pooled samples corresponded to a mixture of both control and treated rat trachea lysates. Samples were analyzed using KinetworksTM KCPS multi-protein immunoblotting analyses with a combination of phospho-site and pan-specific antibodies (Kinexus). The immunoblotting analyses involved probing with mixtures of in-house validated primary antibodies from commercial sources. Immunoblotting was performed using 300 μg of detergent-solubilized lysate proteins as described previously7. The KinetworksTM analysis involves resolution of proteins in a single lysate sample by SDS-PAGE and subsequent immunoblotting overnight at 4°C with panels of up to 3 primary phospho-site-specific antibodies per channel in a 20-lane Immunetics multiblotter. Phospho-site antibodies were primarily obtained from InVitrogen (Carlsbad, CA), Cell Signaling Technologies (Beverly, MA) and Millipore (Temecula, CA). The antibody mixtures were carefully selected to avoid overlapping cross-reactivity with target proteins. Membranes were later rinsed with TBST buffer and then incubated with the relevant horseradish peroxidase conjugated secondary antibody for 45 min at room temperature. The immunoblots were developed with enhanced chemiluminescence (ECL) Plus reagent (Amersham, Arlington Heights, IL) and signals were captured by a Fluor-S MultiImager and quantified using Quantity One software (Bio-Rad, Hercules, CA). Background was less than 100 counts per minute (CPM) for these analyses. From the prescreen of 72 proteins, the 18 most prevalent proteins were identified and evaluated by Western blot as described above in control vs treated pooled rat trachea lysates in order to question whether the exposures altered the amount or phosphorylation of each protein. Protein amount is expressed as normalized CPM’s and % control. Proteins that were considered the most changed not only exhibited significant changes by Western blot analysis, but also represented proteins that displayed a clean band on Western blot.
Results
Pathology
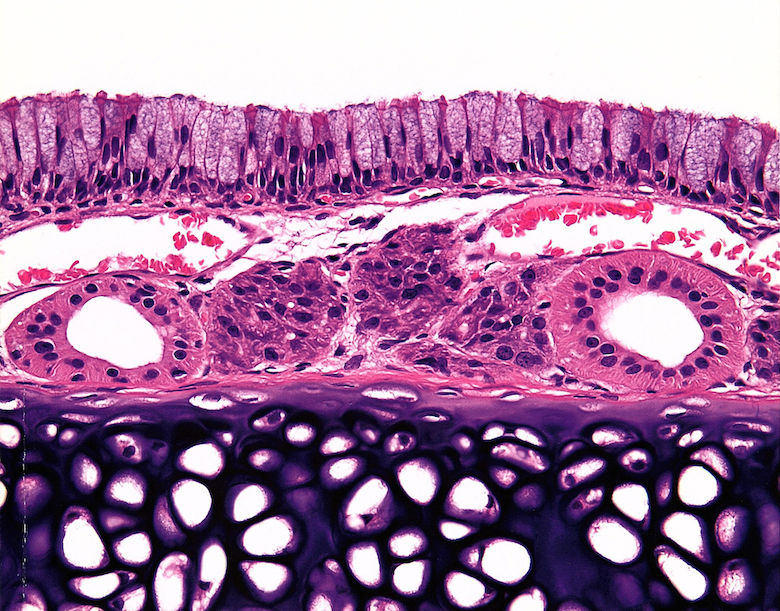
Histopathological changes in smoke-exposed rats consisted of a reduction of tracheal mucosa epithelial layers at 75 and 200 mg TPM/m3 (males and females) and 400 mg TPM/m3 (one female), mucosal hyperplasia at 200 and 400 mg TPM/m3 (males and females), and minimal to mild mucosal erosion in some males and females exposed to 75 and 200 mg TPM/m3 (Table 1). The mucosal hyperplasia at 200 mg TPM/m3 was a focal change in both sexes. The 200 TPM/m3 exposure is of particular interest because it has been related to a heavy smoking pattern in humans which is defined as greater than 23 cigarettes per day8. Tracheal mucosa of controls was comprised of ciliated columnar cells and Clara cells with dome-shaped apical cytoplasm (Fig. 1A & 1E). Minimal focal loss of ciliated cells and minimal focal mucosal thinning was occasionally seen in some male and female controls with a dose-related increase and more generalized occurrence of these effects in smoke-exposed rats. Thinning of tracheal mucosa was more severe in females (Fig. 1F & 1G) than in males (Fig. 1B) while mucosal hyperplasia was more prominent and severe in males (Fig. 1C & 1D) than in females (Fig. 1H). Shortening of cilia and loss of ciliated cells accompanied tracheal mucosal thinning and hyperplasia.
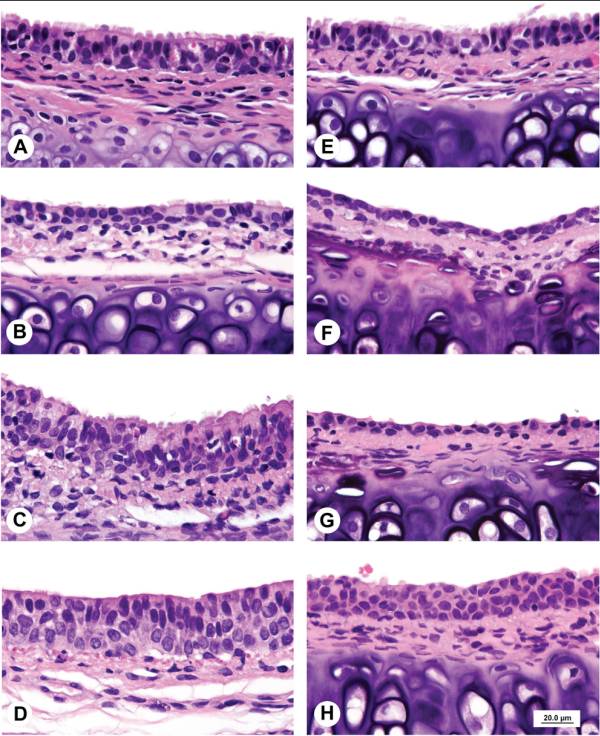
Fig.1. A. Control male. Tracheal mucosa is 2 to 3 cell layers thick with luminal lining comprised of columnar ciliated epithelial cells and Clara cells with dome-shaped apical surfaces. B. Male exposed to 75 mg TPM/m3. Attenuated tracheal mucosa lined by a single layer of cuboidal epithelial cells with shortening and loss of cilia. C. Male exposed to 200 mg TPM/m3. Thickened tracheal mucosa comprised of multiple layers of hyperplastic epithelial cells with decreased ciliated cells and cytoplasmic vacuolation of surface epithelial cells. D. Male exposed to 400 mg TPM/m3. Hyperplastic mucosal epithelial layer consisting of 3 to 4 cell layers with early organization of luminal epithelial cells. E. Control female. Tracheal mucosa is 2 to 3 cells layers thick with luminal lining comprised of tall cuboidal ciliated cells and Clara cells with dome-shaped apices. F. Female exposed to 75 mg TPM/m3. Attenuated tracheal mucosa lined by a single layer of low cuboidal and flattened epithelial cells. G. Female exposed to 200 mg TPM/m3. Markedly attenuated tracheal mucosal surface lined by a single layer of flattened epithelial cells. H. Female exposed to 400 mg TPM/m3. Hyperplastic tracheal mucosa lined by 4 layers of poorly differentiated epithelial cells. Scale bar applies to all figures.
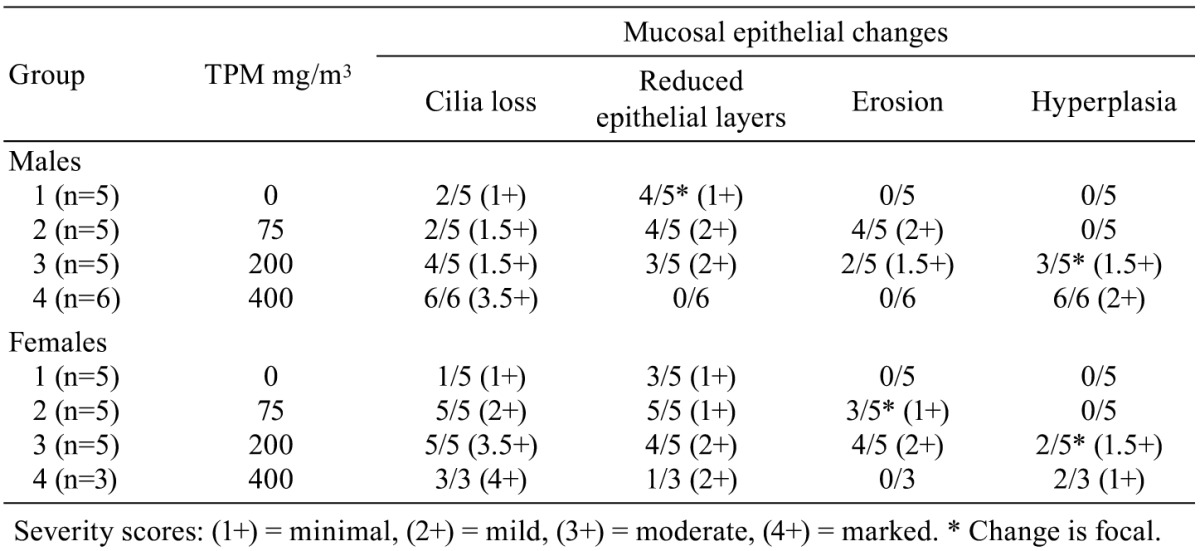
Table 1. Tracheal Changes in Control and Smoke-exposed Rats
Proteomics-antibody microarray analyses
Proteomic analysis of trachea lysates using antibody microarray technology was performed whereby signaling proteins and phosphorylation sites were screened using 800 pan- and phospho-site specific antibodies with lysates from control and cigarette smoke exposed tracheal tissues (Supplemental Table 1: online only). Through a selection process detailed above under Materials and Methods, over 100 antibodies and their target proteins and phospho-sites were subjected to further analyses. Figure 2 details the proteomics analysis scheme.
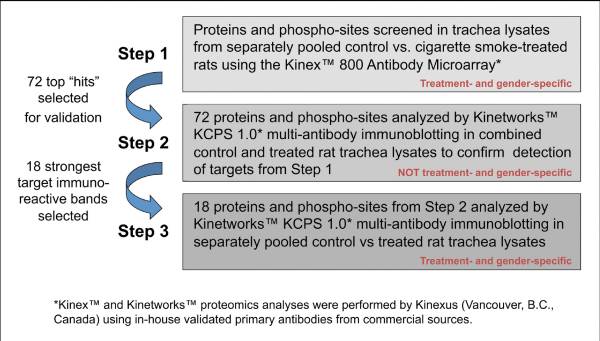
Fig. 2. Schematic detail used for investigation of smoking exposure effects on rat tracheal cell signaling.
Immunoblotting analyses
Immunoblotting analyses of the rat trachea lysates were performed to confirm that antibodies that demonstrated smoke-induced changes with the antibody microarray were in fact specific for their intended target proteins. The proteins that changed most significantly were subjected to additional testing for their ability to show smoke-induced changes in Western blots. The changes of these proteins by Western blot analysis correlated well with the changes by microarray analysis. Figures 3and and44 provide quantitation of the proteins that changed most significantly in rat trachea lysates. Extracellular signal-related kinase (ERK) 1/2, p38 and caspase 5 displayed major increases with smoke exposure in both genders. Upon smoke exposure, p38 increased by 2.5 fold at 200 mg TPM/m3 in both genders and over 2 fold at 400 mg TPM/m3 in the females. In the females, caspase 5 intermediate decreased with smoke exposure while caspase 5 p20 increased. ERK 1/2 increased as much as 2 fold in smoke exposed females compared to controls. PP2Cα (protein phosphatase 2Cα) and PP6C (protein phosphatase 6 catalytic subunit) were increased slightly with some smoke exposures in the females. In the males, caspase 5 intermediate and p20 increased in some smoke exposed groups, while ERK 1/2 increased with all smoke exposures. ERK 2 increased 2 fold at the 200 mg TPM/m3 dose and over 2.5 fold at the 400 mg TPM/m3 dose in males. With smoke exposure, p38 increased in males over 2 fold at the 75 mg TPM/m3 dose and over 2.5 fold at the 200 mg TPM/m3 dose but just over 1.5 fold at the 400 mg TPM/m3 dose. In the males, the PKC (protein kinase C) related kinase PRK2 was slightly elevated at the 75 mg TPM/m3 dose and 200 mg TPM/m3 dose while PP2C/α was decreased with 200 and 400 mg TPM/m3 exposure. PP6C decreased with smoke exposure in the males compared to controls. Western blotting showed that there are differences in both up regulation and down regulation of protein expression and phosphoryation between female and male rats.

Fig. 3. Effects of cigarette smoke exposure on protein expression and phosphorylation in female rat tracheal lysates determined by immunoblotting with specific antibodies. Values are normalized counts per minute (CPM’s) and presented as % control.

Fig. 4. Effects of cigarette smoke exposure on protein expression and phosphorylation in male rat tracheal lysates determined by immunoblotting with specific antibodies. Values are normalized counts per minute (CPM’s) and presented as % control.
Discussion
Cigarette smoke induced tracheal changes including shortening of cilia and loss of ciliated and Clara cells in smoke-exposed rats. In males, cigarette smoke-exposed rat trachea mucosal hyperplasia was prominent at 200 and 400 mg TPM/m3. Cigarette smoke-induced tracheal hyperplasia and tracheal thickening has been reported previously9. Tracheas of male Wistar rats exposed to cigarette smoke for 2 hrs/day for 60 days had loss of cilia, basal cell hyperplasia, goblet cell hyperplasia and an increased number of subepithelial inflammatory cells9. Chronic oxidative stress from cigarette smoke likely contributed to these changes. In contrast to the Isik et al., 20079 study, we did not observe goblet cell hyperplasia or significant inflammation, most likely because of the five-day exposure in our study.
Histopathological effects in our smoke-exposed rats reflect initial irritation and responses in the tracheal mucosa. The cause of the minimal loss of ciliated cells and mucosal thinning in some controls is unknown. However, the dose-related increase in the severity and frequency of these changes and the occurrence of erosions and hyperplasia are consistent with irritation from smoke exposure and a hyperplastic protective and reparative response. Mucosal damage was more severe in females and the reparative hyperplastic response more prominent in males. Thinning of the tracheal mucosa in lower exposure groups would be the expected consequence of apoptosis and loss of respiratory epithelium approximately a day or two before study termination at five days. The absence of inflammation in the trachea is consistent with loss of epithelium due to apoptosis rather than necrosis. Reparative mucosal hyperplasia was present in tracheas of rats exposed to higher concentrations of smoke, indicating that initial loss of epithelium occurred earlier at the higher, presumably more toxic, exposure levels allowing sufficient time to begin replacement of lost mucosal epithelium prior to study termination.
In the present study, cigarette smoke exposure generally induced increases in extracellular signal-related kinase (ERK)1/2, p38 and caspase 5 in rat trachea lysates. These proteins regulate a spectrum of cellular functions including apoptosis, inflammation, proliferation, differentiation, and stress pathways10,11,12,13. The mitogen-activated protein (MAP) kinases, ERK, p38 and JNK (c-Jun NH2-terminal kinases) are implicated in various disease processes. Stress stimuli activate p38 and JNK, while ERK helps regulate cell proliferation, differentiation, and survival10,14,15. ERK 2 and ERK1/2 were generally increased in tracheal lysates from smoke-exposed rats and are believed to be regulating a proliferative signal to stimulate repair of damaged tracheal mucosa. The morphological correlate of the ERK proliferative signal is the observed tracheal mucosal hyperplasia present in the 200 mg males and 400 mg males and females. The fact that elevated ERKs levels did not consistently correlate with a proliferative morphologic response may reflect a balance between the negative effects of ERK1 on the positive proliferative function of ERK216. Similar to our proteomics results, cigarette smoke increased ERK1/2 phosphorylation in airway lining cells and alveolar macrophages in mice exposed for 10 days and 6 months17. ERK1/2 also significantly increased in lung tissues from emphysema patients compared to controls17. Cigarette smoke activated ERK 1/2 in human bronchial epithelial cells18 and significantly increased phosphorylation of ERK 1/2 in human aortic smooth muscle cells19. In lung lysates from Fischer rats from the same exposure regimen as in the present study, ERK2 increased 79% in 75 mg TPM/m3 females and 41% in 200 mg TPM/m3 males6. Taken together, cigarette smoke has a prominent modulatory effect on ERK protein levels in respiratory system tissues. ERK plays an important role as a mediator of the proliferative signal20, and serves a role in regeneration and protection against cellular injury21 as well as becoming activated by cigarette smoke induced apoptosis12.
p38 is responsive to stress stimuli and plays a role in apoptosis and cellular differentiation. The initial loss of tracheal mucosal cells followed by a hyperplastic response at the higher smoke exposure levels is consistent with initial apoptotic cell loss and subsequent cellular differentiation accompanying the reparative hyperplastic response. Similar to our p38 results, sidestream cigarette smoke stimulated p38 phosphorylation in human endothelial cells22. Because activation of p38 can be induced by airborne pollutant particulate matter23 our p38 increases may reflect a generalized response to particulates associated with smoke exposure.
The initial thinning of the tracheal mucosal in smoke-exposed rats likely reflects an apoptotic loss and in addition to p38, two other protein hits, caspase 5 and PP6C, may be contributing to this effect. Caspase 5 mediates apoptosis and is an inflammatory caspase regulated by lipopolysaccharide and interferon γ24. Caspase 5 p20 is a fully cleaved or activated form of capase 5 and increased in tracheal lysates (200 and 400 mg TPM/m3 in both genders vs controls) while in lung lysates from Fischer rats from the same study, caspase 5 decreased 55% in 75 mg TPM/m3females and 63% in 200 mg TPM/m3 males6. Caspase 5 intermediate domain (30 kDa) does not fully lead to a similar expression of p20 caspase 5 in tracheal lysates, especially not in males.
Changes in PRK 2 and the phosphatases PP2C/α and PP6C were similar to control values except for PP6C, which decreased in smoke exposed males vs. controls. PP6C is involved in cell cycle regulation and restricts cancer cell growth25. PP6C is involved in the DNA damage response and causes sustained activation of H2AX26. PP6C regulates apoptosis and decreases in PP6C have been related to a high rate of apoptosis27. The PP6C decrease in smoke exposed males is consistent with apoptotic loss of tracheal mucosal epithelium at the 75 mg TPM/m3 dose and may reflect apoptosis associated remodeling as part of the reparative hyperplasia seen at 200 and 400 mg TPM/m3.
The most pronounced smoke-altered proteins in rat tracheal lysates regulate apoptosis, stress response, proliferation, differentiation, survival, and inflammation. ERK 1/2, p38 and caspase 5 were the proteins most increased in trachea lysates and are associated with apoptosis regulation. Cigarette smoke is a strong apoptotic inducer in the respiratory tract28. Cilia loss and hyperplasia observed in exposed animals would likely contribute to lack of mucociliary clearance and other alterations in tissue function. The initial hypothesis that smoke-altered proteins in rat tracheal isolates reflect pathways involved in stress response, proliferation, apoptosis and inflammation was supported in this study with the exception that there was no significant inflammation observed. Cilia loss, mucosal thinning, and hyperplasia of the tracheal mucosa are phenotypic responses consistent with the major alteration of the proteome identified in this study. Biomarker identification such as the ones revealed in this study may allow development of screening assays to reveal individuals more susceptible to the effects of toxins.
Acknowledgments
We thank Dr. Steven Pelech, Catherine Sutter, Litsa Blanis and Kinexus staff for performance of the proteomics screen and for helpful scientific comments, and Nancy Harris and the EPL staff for immunostaining.
References
1. Romet-Haddad S, Marano F, Blanquart C, Baeza-Squiban A. Tracheal epithelium in culture: a model for toxicity testing of inhaled molecules. Cell Biol Toxicol. 8: 141–150 1992.
2. Carter CA, Doherty MM, Rusnak DW, Nettesheim P, Ferriola PC. Alterations in the localization of F-actin, fibronectin, and thrombospondin occur prior to neoplastic transformation in rat tracheal epithelial cells. Exp Cell Res. 212: 141–150 1994.
3. Newland N, Baxter A, Hewitt K, Minet E. CYP1A1/1B1 and CYP2A6/2A13 activity is conserved in cultures of differentiated primary human tracheobronchial epithelial cells. Toxicol In Vitro. 25: 922–929 2011.
4. Turetz ML, O’Connor TP, Tilley AE, Strulovici-Barel Y, Salit J, Dang D, Teater M, Mezey J, Clark AG, Crystal RG. Trachea epithelium as a “canary” for cigarette smoking-induced biologic phenotype of the small airway epithelium. Clin Transl Sci. 2: 260–272 2009.
5. Battershill JM. The multiple chemicals and actions model of carcinogenesis. A possible new approach to developing prevention strategies for environmental carcinogenesis. Hum Exp Toxicol. 24: 547–558 2005.
6. Carter CA, Misra M, Pelech S. Proteomic analyses of lung lysates from short-term exposure of Fischer 344 rats to cigarette smoke. J Proteome Res. 10: 3720–3731 2011.
7. Pelech S, Jelinkova L, Susor A, Zhang H, Shi X, Pavlok A, Kubelka M, Kovarova H. Antibody microarray analyses of signal transduction protein expression and phosphorylation during porcine oocyte maturation. J Proteome Res. 7: 2860–2871 2008.
8. Finch GL, Nikula KJ, Chen BT, Barr EB, Chang IY, Hobbs CH. Effect of chronic cigarette smoke exposure on lung clearance of tracer particles inhaled by rats. Fundam Appl Toxicol. 24: 76–85 1995.
9. Işik AC, Yardimci S, Guven C, Avunduk MC, Civelek S. Morphologic alteration induced by short-term smoke exposure in rats. ORL J Otorhinolaryngol Relat Spec. 69: 13–17 2007.
10. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 410: 37–40 2001.
11. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 14: 10–22 2007.
12. Kuo WH, Chen JH, Lin HH, Chen BC, Hsu JD, Wang CJ. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact. 155: 31–42 2005.
13. Park JW, Yoon JY, Kim YJ, Kyung SY, Lee SP, Jeong SH, Moon C. Extracellular signal-regulated kinase (ERK) inhibition attenuates cigarette smoke extract (CSE) induced-death inducing signaling complex (DISC) formation in human lung fibroblasts (MRC-5) cells. J Toxicol Sci. 35: 33–39 2010.
14. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 12: 14–21 2002.
15. Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 68: 320–344 2004.
16. Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 5: 14 2006.
17. Mercer BA, Kolesnikova N, Sonett J, D’Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 279: 17690–17696 2004.
18. Petecchia L, Sabatini F, Varesio L, Camoirano A, Usai C, Pezzolo A, Rossi GA. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest. 135: 1502–1512 2009.
19. Xu CB, Lei Y, Chen Q, Pehrson C, Larsson L, Edvinsson L. Cigarette smoke extracts promote vascular smooth muscle cell proliferation and enhances contractile responses in the vasculature and airway. Basic Clin Pharmacol Toxicol. 107: 940–948 2010.
20. Lloyd AC. Distinct functions for ERKs? J Biol. 5: 13 2006.
21. Kunduzova OR, Bianchi P, Pizzinat N, Escourrou G, Seguelas MH, Parini A, Cambon C. Regulation of JNK/ERK activation, cell apoptosis, and tissue regeneration by monoamine oxidases after renal ischemia-reperfusion. FASEB J. 16: 1129–1131 2002.
22. Low B, Liang M, Fu J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J Pharmacol Sci. 104: 225–231 2007.
23. Watterson TL, Hamilton B, Martin R, Coulombe RA., Jr Urban particulate matter causes ER stress and the unfolded protein response in human lung cells. Toxicol Sci. 112: 111–122 2009.
24. Lin XY, Choi MS, Porter AG. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem. 275: 39920–39926 2000.
25. Bastians H, Ponstingl H. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J Cell Sci. 109(Pt 12): 2865–2874 1996.
26. Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 30: 1368–1381 2010.
27. Kajihara R, Fukushige S, Shioda N, Tanabe K, Fukunaga K, Inui S. CaMKII phosphorylates serine 10 of p27 and confers apoptosis resistance to HeLa cells. Biochem Biophys Res Commun. 401: 350–355 2010.
28. D’Agostini F, Balansky RM, Izzotti A, Lubet RA, Kelloff GJ, De Flora S. Modulation of apoptosis by cigarette smoke and cancer chemopreventive agents in the respiratory tract of rats. Carcinogenesis. 22: 375–380 2001.