We foresee cancer research as an increasingly logical science, in which complexities are manifestations of a small set of underlying organizing principles. (Hanahan and Weinberg, 2011)
1. INTRODUCTION
1.1. Overview
Cancer is a major cause of debilitation and death in humans and animals. Cancer develops as a function of age, environment, diet, and genetic makeup, whether in man or animals. As humans reach their sixth decade, they face an exponentially increased risk for developing cancer. There is a similar window of increased susceptibility to chemically-induced neoplasms in rodents treated with either a single or chronic exposure(s) to carcinogen(s). Identifying potential human carcinogens in rodent bioassays has been a major focus of the field of toxicologic pathology.
It is estimated that 5% of human cancers are caused by viruses, 5% by radiation, and the remaining 90% by chemicals. Of these, an estimated 30% are caused by the use of tobacco products and the rest by chemicals associated with diet, lifestyle, and the environment. The importance of chemical products in the etiology of cancer is reflected in the fact that up to 8% of all human cancers are related to occupational chemical exposure. All chemical carcinogens, or their derivatives, are highly reactive electrophiles, which have electron-deficient atoms that can react with nucleophilic, electron-rich sites in the cell. Deoxyribonucleic acid (DNA), in particular, is made up of an array of nucleophilic centers at which these DNA-damaging agents can form adducts through one or more covalent bonds.
To date, approximately 6 million chemicals have been identified and registered with the chemical abstracts services. Of these, more than an estimated 50,000 are used regularly in commerce and industry. Less than 2000, however, have been examined for their carcinogenic potential.
According to the International Agency of Research on Cancer (IARC), the United States Environmental Protection Agency (EPA), and the United States National Toxicology Program’s Report on Carcinogens (RoC), there are more than 100 known human carcinogens (Table 5.1) and virtually all of them also cause cancer in animals. Animal and human cancers are fundamentally similar and frequently share morphological, biological, and molecular biological features. In fact, approximately 30% of human carcinogens were first identified in animal studies. Two Japanese pathologists, Yamigawa and Ichikawa, are credited with the original demonstration that a chemical could produce cancer in animals. With chronic exposure of the skin (pinna) to coal tars, rabbits developed squamous cell carcinomas, some of which metastasized. These findings in 1918 confirmed Percival Pott’s strong epidemiological observations in 1775 of increased rates of cutaneous scrotal cancer in chimney sweeps and demonstrated that chronic exposures were necessary for the induction of some cancers.
Chronic inflammation and infectious agents have also been implicated in the development of a number of human and animal cancers. Nobel laureates Drs Robin Warren and Barry Marshall were credited with determining that infection with Helicobacter pylori was a common cause of gastric inflammation and ulcers in man. Some skeptical scientists were not convinced until Dr Marshall developed gastritis soon after drinking a Petri dish of the bacteria.
In subsequent years it was shown that chronic helicobacter gastritis is associated with the development of gastric lymphomas and carcinomas, and thereby Helicobacter pylori is listed as a human carcinogen. Some of the lymphomas appear reversible and regress after treatment with antibiotics. Certain strains of mice (including A/JCr and B6C3F1/N) with chronic Helicobacter hepaticus hepatitis develop signifi- cantly higher rates of liver cancer compared to uninfected controls. Helicobacter infections may confound interpretation of the results of cancer occurrence in animal bioassays or human epidemiological studies.
Our understanding of cancer biology is evolving at a rapid rate. Conceptual views of carcinogenesis are formed by the piece-by-piece discovery of key elements of the complex biological puzzle that this disease entails. This pieceby-piece discovery includes early findings of the evidence of clonal evolution of cancer, the Knudsen two-hit hypothesis progression from benign to malignant growth, discovery of oncogenes and tumor suppressor genes, the somatic mutation theory, the Fearon-Vogelstein multistep colon cancer mutation model, mutator phenotype, and the cancer stem-cell theory.
With the advent of new technologies in molecular analysis, such as gene expression profiling, networks, microRNAs, gene discovery, and pathway analysis, carcinogenesis is proving to be much more complex than being simply a clonal evolution of a cell that sustained two genetic “hits” by a carcinogen. The current multistep model of carcinogenesis involves at least 80 cancer gene mutations or alterations, about a dozen of which are “drivers” of the cancer growth processes. The hallmarks of carcinogenesis (Hanahan and Weinberg, 2011) include genetic alterations involved in:
- sustaining proliferative signaling;
- evading growth suppressors;
- resisting cell death;
- enabling replicative immortality;
- inducing angiogenesis;
- activating invasion and metastasis;
- reprogramming energy metabolism; and
- evading immune destruction.

1.2. Background
It is still accepted today that cancer originates in a single cell and develops through the clonal proliferation of their progeny. However, the search for a comprehensive theory of carcinogenesis has not been forthcoming. Investigation into the two predominant and antagonistic theories: humoralist and cellular, dominated most of the second half of the 19th century.
The humoralist view regarded cancer as originating from certain hereditary characteristics of the individual associated with susceptibility to contract the disease. Cellular pathologists such as Muller and Virchow, however, argued that cancer was related to a form of chronic irritation. This latter view was supported by experimental studies in mouse skin where wounding seemed to play a tumorigenic role. Over the next century, thousands of chemicals have been shown to transform cells in vitro and to be carcinogenic in animals. Those that are known to be carcinogenic in humans are many times fewer (see Tables 5.1 and 5.2).
Some of the most potent carcinogens have been extracted from fossil fuels or are synthetic products created by industry. However, a variety of occupational causes of cancer had been documented prior to the Industrial Revolution. In 1531, Paracelsus had described the “mala metallorum” among miners for silver and other metals, including uranium. This observation was later interpreted as radiation-induced lung cancer. In 1775, Pott attributed scrotal skin cancers to prolonged exposure to soot in chimney sweeps. A few years later, on the basis of this observation, the Danish Chimney Sweeps Guild ruled that its members must bathe daily. No public health measure since that time has so successfully controlled a form of cancer.
Cancers related to industrial activity were reported early in the industrial age in several locations with increasing frequency. Examples include the “aniline” bladder cancer related to the systemic effects of naphthylamines and the “paraffin cancer” caused by exposure to shale oil. The interest in industrial carcinogens coexisted with the realization that a definitive cause–effect relationship existed between tobacco and cancer.
Early reports by Hill in 1761, which called attention to the association of “immoderate use of snuff” and the development of “polypusses,” later received experimental confirmation by Roffo in 1931 when he induced skin cancer in rabbits by painting with tobacco-derived tar. However, tobacco tar was shown to be only weakly carcinogenic in experimental models. Thus, a serious effort was undertaken to identify potential carcinogens using the mouse skin experimental model.
Polycyclic aromatic hydrocarbons were the initial target of research. Benzo[a]pyrene, the most potent carcinogenic agent of tar, is present in the environment as a result of cigarette smoke and automobile exhaust fumes. It became the most intensely studied chemical carcinogen because of the belief that its chemical structure was related to that of steroid hormones. Steroidal hormones were at the time the only endogenous compounds reported to induce tumors in mice and humans. It was shown later that compounds with many different chemical structures could induce neoplasia. It also became clear that age and individual differences were contributing factors in the susceptibility to cancer, and these differences, presumably of genetic origin, could be inherited.

1.3. Initiation, Promotion, and Progression
In early studies, it was observed that a long latent period could elapse from exposure to carcinogens to the development of cancer. In 1941, Rous and Kidd painted the skin of rabbits and found that if this painting were interrupted, tumors would disappear only to reappear if the application of tar was re-established. It seemed, therefore, that a reversible process was taking place in those cells that did not attain the complete neoplastic state.
These cells had undergone what Rous called initiation. Further development of tumors would then require what was termed promotion, the process by which the initiated cell expands clonally into a detectable cell mass that is either benign or preneoplastic. Finally, cells must undergo additional changes in their progression to a malignant neoplasm. Today it is known that for a normal cell to evolve into a malignant one, heritable changes involving multiple, independent genes are required. This “multi-hit” model is consistent with the incidence rates of cancer that increase exponentially with age.
The concepts of initiation and promotion were first described during experiments on mouse skin carcinogenesis, but have since been applied to a variety of other tissues and species. During the initiation phase of chemical carcinogenesis, a normal cell undergoes an irreversible change characterized by an intrinsic capacity for autonomous growth. This capacity for autonomous growth remains latent for weeks, months, or years, during which time the initiated cell may be phenotypically indistinguishable from other parenchymal cells in that tissue.
Spontaneous initiation refers to a situation in which the exact agent responsible for the initiating event is unknown. One possibility that can lead to spontaneous initiation would be a situation wherein there is infidelity in the action of DNA polymerase during normal cellular division or during DNA repair. Initiation operationally implies that there is alteration to cellular DNA at one or more sites in the genome. Such alteration represents a mutational event, which can be hereditary.
Metabolic activation of a carcinogen to its chemically reactive products and their subsequent reaction with cellular targets (e.g., DNA bases) occurs within a few hours of exposure. Most tissues have the ability to repair this damage over a period of days or weeks. Currently accepted dogma suggests that the chemically damaged DNA, if not first repaired by normal cellular processes, is converted to a stable biological lesion (mutation, chromosomal rearrangement, etc.) during DNA replication. Thus, if a round of cell replication occurs before the DNA damage is repaired, the lesion in the DNA is regarded as “fixed.” This phenomenon may explain the high frequency of neoplasms in proliferating tissue where there is an intrinsically high rate of cell turnover coincident with exposure to a DNA-damaging agent.
In contrast to the step of initiation, the conversion of an initiated cell to a fully malignant neoplasm is usually a prolonged process, lasting months in animals and years in humans. Based on the hypothesis that most initiators are mutagenic or genotoxic, a battery of short-term in vitro and in vivo mutagenicity tests have been developed to permit the detection of chemicals with potential initiating activity. Identification of initiating agents is especially important because of the irreversible and hereditary nature of the alterations that occur during initiation. While useful when positive results are obtained, the predictive ability of short-term mutagenicity tests for the ultimate carcinogenic potential of xenobiotics is not absolute.
Exposure of experimental animals to chemicals with initiating activity may ultimately result in the induction of multiple neoplasms in a given tissue. Each individual neoplasm is often found to be monoclonal in origin, having arisen from a single initiated cell. Application of techniques such as identification of cell surface immunoglobulin markers and glucose-6-phosphatase dehydrogenase variants, restriction fragment length polymorphisms, cytogenetic studies, single-cell transplantation studies, and identifi- cation of chromosome inactivation mosaics has permitted identification of the monoclonal and polyclonal nature of individual neoplasms.
The vast majority of human and animal neoplasms studied to date are of monoclonal origin. There are several salient characteristics of initiation. It can occur following a single exposure to a known carcinogen. Changes produced by the initiator may be latent for weeks or months, and are considered irreversible. The interval between initiation and promotion may be as long as 1 year in mouse skin-painting studies.
Several lines of evidence indicate that initiation is additive and that the yield of neoplasms is dose-dependent. Increasing the dose of initiator increases the incidence and multiplicity of resulting neoplasms and shortens the latency to manifestation of neoplasms. Because the initiating event must be “fixed” by a round of cell proliferation, it becomes obvious that initiation is dependent on the cell cycle.
Finally, there is no readily measurable threshold dose for maximum and minimum responses to initiators. Unrealistically large numbers of animals would be required to demonstrate minimum responses, and confluence of multiple neoplasms following high doses precludes accurate quantitation of the neoplastic response. Properties of initiators are summarized in Table 5.3.

Initiators interact with host cellular macromolecules and nucleic acids in specific patterns, typically involving the generation of reactive electrophiles, esters, or free radicals that bind covalently to nucleophilic sites in critical cellular macromolecules. The majority of known carcinogens have both initiating and promoting activity and can thus induce neoplasms rapidly and in high yield when given repeatedly. When given at sufficiently low single doses, even a complete carcinogen may act as a “pure” initiator requiring subsequent promotion for the detection of any neoplasms. Under such circumstances, the agent can be regarded operationally as an “incomplete carcinogen.” A cell that has undergone the irreversible change that permits its ultimate neoplastic transformation may be phenotypically indistinguishable from adjacent normal parenchymal cells. However, when stimulated properly, it has an intrinsic capacity for autonomous growth.
Promotion is classically considered that portion of the multistep carcinogenic process where specific agents, known as promoters, enhance the development of neoplasms from a background of initiated cells. The promoting agents themselves are so classified only in an operational sense.
A promoter is typically given at some time after chemically-induced or fortuitous initiation, and the doses of agent used are insufficient to produce cancer without prior initiation. It should be appreciated that when classical promoters are administered at sufficiently high doses and for prolonged intervals, neoplasia can occur without evidence of prior initiation. Under these conditions, a promoting agent must be considered a complete carcinogen unless fortuitous initiation from background radiation, dietary contaminants, environmental toxins, and so on is believed to have occurred. However, under typical experimental conditions commonly employed in short- and medium-term initiation–promotion experiments, neoplasia does not typically occur in animals that have not been previously initiated. The temporal sequence of promoter administration is critical to the operational definition of promotion. The agent must be administered after initiation and cause enhancement of the neoplastic process to be considered a promoter. If an agent is given simultaneously with an initiator and results in enhancement of the development of neoplasms, it is regarded as a co-carcinogen rather than a promoter.
While some promoters such as phorbol esters are co-carcinogenic, not all promoters (e.g., phenobarbital, phenol) possess co-carcinogenicity and, conversely, not all co-carcinogens are promoters. Under these same conditions of simultaneous administration, a diminution in the neoplasm response is considered evidence of anti-carcinogenic activity. Thus, several rodent liver tumor promoters, which are active when given after a variety of initiators, prevent or delay the development of liver neoplasms when added to diets along with an active carcinogen.
Finally, reversing the order of administration by giving a known promoter prior to an initiator may prevent the expression of carcinogenic activity on the part of the initiator. While upper and lower thresholds have been demonstrated experimentally for promoters, some consider that, in an absolute sense, it is statistically impossible unequivocally to prove or disprove the existence of thresholds for promoters for much the same reasons that this cannot be done for initiators. One can never be certain that an apparent no observable effect level (NOEL) would indeed be without effect if a sufficiently large enough number of animals were used.
Promoters include agents such as drugs, plant products, and hormones that do not interact directly with host cellular DNA (are not genotoxic) but somehow influence the expression of genetic information encoded in the cellular DNA (e.g., epigenetics). It has been suggested that promoting agents may cause gene repression and derepression in cells. Some experimental evidence suggests that the regulation of gene expression is unique to the nature of the promoting agent administered. Some promoters are believed to produce their effect by interaction with receptors in the cell membrane, cytoplasm, or nucleus (e.g., hormones, dioxin, phorbol ester, polychlorinated biphenyls). Alternatively, some hydrophilic and hydrophobic promoting agents exert their effect through their molecular orientation at cellular interfaces. Other promoters are mitogenic, stimulating DNA synthesis and enhanced cell proliferation. This may occur directly or, alternatively, indirectly by targeting cells with a shortened G1 phase, thereby giving them a selective proliferative advantage. At least in tissue culture, some promoters have been shown to inhibit intercellular communication (metabolic cooperation). In some situations, metabolism appears to play little role in the action of promoters.
Experimental evidence suggests that the molecule as a whole may exert the promotional effect and that the molecular configuration determines the activity of the agent. When promoter metabolism does occur, it typically results in inactivation of the agent. Possible exceptions include d-limonene and trimethyl pentane (unleaded gasoline), which are promoters of renal tumors in rats.
Promoters appear to have relatively high tissue specificity. Thus, phenobarbital functions as a promoter for rodent liver neoplasia but not in the urinary bladder. In contrast, 12-0-tetradecanoylphorbol-13-acetate is a potent skin and forestomach neoplasm promoter, but has no appreciable activity in the liver. Antioxidants such as 3-tert-butyl-4-methoxyphenol and 2,6-ditert-butyl-4-methoxyphenol may act as promoters in one organ, anti-promoters in another organ, and have no effect in a third organ. Thus, the practical definition of a promoter must include the designation of the susceptible tissue.
Tumor promotion may be modulated by several factors, such as age, sex, diet, and hormone balance. The correlation of increased rates of breast cancer in women following a “western” life style has implicated meat and fat consumption as playing an important role in breast cancer development. Experimental demonstration of the role of a high-fat diet in the promotion of mammary cancer in rats exposed to the mammary carcinogen dimethylbenzanthracene (DMBA) has been documented.
Similarly, bile acids, as modulated by fat consumption, are known promoters of rat liver carcinogenesis. Age- and sex-associated modulations in hormonal levels of estrogens, progesterone, and androgens have been implicated as potential promoters of breast cancer on the basis of epidemiology studies in humans. Experimental studies have shown repeatedly that these hormones, in addition to pituitary prolactin, serve to promote mammary cancer in rats initiated with mammary carcinogens.
Some promoters cause hyperplasia and/or inflammation. This is particularly true in skin initiation–promotion studies using phorbol esters as promoters, but is also seen in hyperplasia of hepatocytes following treatment with mitogenic agents such as phenobarbital. In the rodent liver, phenobarbital causes a transient hyperplasia of hepatocytes. It should be remembered that some materials could cause hyperplasia and inflammation but be without promoting effects.
This observation has led some investigators to consider that the ability to stimulate DNA synthesis and cell division and the ability to induce inflammation are essential but not sufficient properties of promoters. Properties of promoters are summarized in Table 5.3. It has been suggested and confirmed experimentally that the process of promotion can be divided into at least two stages in the mouse skin-painting initiation–promotion model. The two-stage promotion model is based on the stimulation of basal cell hyperplasia (dark basal keratinocytes) in stage 1 of promotion followed by enhancement of cell proliferation in stage 2.
In the classical mouse skin carcinogenesis model, a phorbol ester binds to a membrane receptor, induces protein kinase C (PKC), and is effective with only one application, the process being at least partially irreversible, in the first stage of promotion. Weak or non-promoting agents such as mezerein are effective as second-stage promoters, but require multiple applications and do not always have receptor-binding properties. Similar multistage promotion has not yet been demonstrated in other experimental carcinogenesis model systems.
Progression is that part of the multistep neoplastic process associated with the development of an initiated cell into a biologically malignant cell population. In common usage, progression is used frequently to signify the stages whereby a benign proliferation becomes malignant or where a neoplasm develops from a low grade to a high grade of malignancy. During progression, neoplasms show progressively increased invasiveness, develop the ability to metastasize, and have alterations in biochemical, metabolic, and morphologic characteristics.
Tumor cell heterogeneity is an important characteristic of tumor progression. Expression of this heterogeneity includes antigenic and protein product variants, ability to elaborate angiogenesis factors, emergence of chromosomal variants, development of metastatic capability, altered metabolism, and decreased sensitivity to radiation. The development of intraneoplastic diversity may come about as a consequence of genetic change such as loss of polymorphic restriction fragments in DNA of malignant tumors or similar random processes such as additional genomic “hits” by genotoxic agents.
Alternatively, the heterogeneity observed in tumor progression may be generated by epigenetic, regulatory mechanisms operative as a continuation of the process of promotion. More than likely, genetic and epigenetic events subsequent to initiation operate in a non-mutually exclusive manner during progression, possibly in an ordered cascade of latter events superimposed on earlier events.
The most plausible mechanism of progression invokes the notion that during the process of tumor growth there is a selection that favors enhanced growth of a subpopulation of the neoplastic cells. In support of this mechanism is the observation of increased phenotypic heterogeneity that is observed in malignant versus benign neoplastic proliferations. Presumably a variety of subpopulations arise and it is only a matter of time before the emergence of a subpopulation with more malignant biological characteristics or at least a differential growth advantage. This can occasionally be observed during early stages of experimental hepatocarcinogenesis when phenotypically distinguishable neoplasms can arise within existing foci of altered hepatocytes.
In addition, serial transplantation of tumors frequently shows an enhancement of malignant properties associated with increased numbers of passages. Here there is a presumed selection for more rapidly growing subpopulations because the subsequent serial passage is typically dictated by the size of the transplant growth in the recipient.
Associated with progression is the development of an increased degree of karyotypic instability and of aneuploidy. This latter phenomenon may be related to the not infrequent observation of abnormal mitoses in malignant neoplasms. Finally, chromosomal rearrangement is associated with several clinically malignant neoplasms, especially leukemias. It is probable that such rearrangements are a consequence of karyotypic instability and that apposition of critical portions of genes upstream or downstream from genomic enhancers or derepressors imparts a proliferative advantage and a metastatic capability to affected cells within an evolving neoplastic proliferation.
Distinction between tumor promotion and tumor progression is not readily discernible in the routine histopathologic evaluation of neoplasms. In fact, the distinction may be somewhat academic in that promotion may be considered part of the process of progression. In both situations the critical event is accentuated growth.
Distinguishing progression from promotion is the presence of structural genomic alterations in the former and the absence of definable structural changes in the genome of the latter. Both structural genomic changes and biochemical changes associated with tumor progression cannot be defined by conventional histopathology. Emerging technologies centered on histochemistry, immunocytochemistry, and in situ hybridization to identify products of proto-oncogenes and activated oncogenes offer promise to help distinguish various stages of progression in the evolution from benign to malignant neoplasms.
1.4. Preneoplasia
Most neoplasms are believed to be derived from the clonal proliferation of a single initiated cell. Usually at some point early in the clonal expansion, the differentially proliferating cells may become phenotypically distinguishable from the surrounding normal parenchyma. Although such lesions may not as yet have sufficient characteristics to qualify as neoplasms, their recognition has led many to regard them as “preneoplastic.” According to the multistage model of carcinogenesis, there is a morphological continuum from hyperplasia/preneoplasia to adenoma and carcinoma, which is reflected in the pattern observed in tumor types.
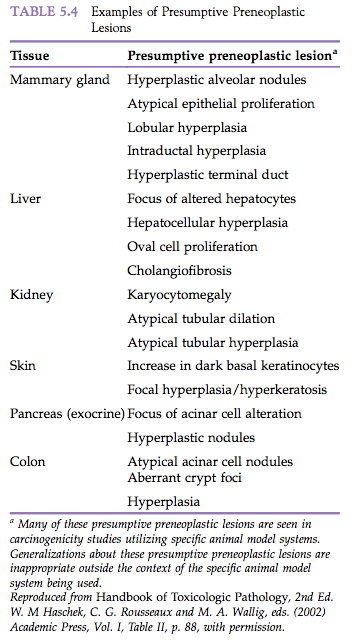
Patients or animals with preneoplastic lesions are at increased risk of developing neoplasia at the tissue site in which the preneoplasia is present. The preneoplastic lesions themselves are believed to progress to neoplasia, although unequivocal proof for this is difficult to document. Examples of preneoplasia from human medicine include leukoplakia of the oral cavity or vulva, senile keratosis, and xeroderma pigmentosum. Numerous putative preneoplastic lesions have been identified in laboratory animals (Table 5.4).
A well-studied example of preneoplasia is seen in experimental studies of liver neoplasia using rats exposed to potent hepatocarcinogens. The initial change detected in liver tissue consists of foci of cellular alteration. These foci consist of nests or islands of altered hepatocytes that differ phenotypically from adjacent normal hepatocytes due to the tinctorial quality of the cytoplasm. The foci of altered hepatocytes (FAH) often precede the development of hepatocellular neoplasms.
There are several phenotypic types of FAH, including eosinophilic, basophilic, clear cell, and mixed, and the ultimate neoplasms may resemble them phenotypically. Because of their consistent production by known hepatocarcinogens and their temporal relationship with ultimate neoplasia, FAH are regarded operationally as preneoplastic lesions. Because the number of neoplasms eventually generated represents a very small proportion of the number of foci produced (estimates range from 1 neoplasm for every 1000 to 10 000 foci), conservative pathologists regard the foci as “putatively preneoplastic.”
The exact significance of FAH is unknown. Empirical observations speak both for and against their role in oncogenesis. On the one hand, FAH occur spontaneously in aging untreated rats that have a negligible background incidence of hepatic neoplasia. Also, in some experimental models, the stability or persistence of foci is dependent on continued exposure to a xenobiotic agent or feed ingredient. On the other hand, the consistent induction of foci by known hepatocarcinogens and the occasional observation of “neoplasia within a focus” suggest that these putatively preneoplastic lesions are related to hepatic neoplasia. This finding may simply reflect that additional critical genetic perturbations to undergo neoplastic transformation have occurred. Despite the absence of definitive proof that hepatic foci of cellular alteration are precursor lesions for hepatic neoplasms, several investigators have identified chemicals as having hepatocarcinogenic activity based only on induction of foci by these chemicals. It has been proposed that regulatory decisions regarding the production, distribution, and use of chemicals can be based on the observation that they induce FAH in rat carcinogenicity studies.
1.5. Hypertrophy
While not technically a proliferative change, hypertrophy deserves mention because it is sometimes diagnosed incorrectly as hyperplasia (Morphologic Manifestations of Toxic Cell Injury, Chapter 4). Hypertrophy and hyperplasia may occur together, and several factors link hypertrophy to rodent hepatocarcinogenesis (Table 5.5). Hypertrophy is under various regulatory controls, and thus is limited in amount and duration. Hypertrophy may be classified in a manner similar to how hyperplasia is classified. Compensatory or adaptive hypertrophy represents a physiological response to a stimulus, such as is seen with muscle hypertrophy subsequent to prolonged exercise or in enzyme induction in the liver following exposure to chemical inducers such as phenobarbital. Combined with degenerative changes, such as necrosis and vacuolization, hepatocyte hypertrophy is associated with the development of hepatocellular neoplasms in rats and mice.
Exposure of animals to liver enzyme inducers results in a signature of toxicological changes characterized by an increase in liver weight, hepatocellular hypertrophy, cell proliferation, and (frequently in chronic [2-year] studies) hepatocarcinogenesis (Liver and Gallbladder, Chapter 45). Hepatic enzyme induction is generally an adaptive response associated with increases in liver weight, induction of gene expression, and morphological changes in hepatocytes. The additive growth and functional demands that initiated the response to hepatic enzyme induction cover a wide range of stimuli, including pregnancy and lactation, hormonal fluctuations, dietary constituents, infections associated with acute phase proteins, as well as responses to exposure to xenobiotics. Common xenobiotic enzyme inducers trigger pathways involving the constitutive androstane receptor (CAR), the peroxisome proliferator-activated receptor (PPAR), the aryl hydrocarbon receptor (AhR), and the pregnane-X-receptor (PXR). Liver enlargement in response to hepatic enzyme induction is typically associated with hepatocellular hypertrophy and often with transient hepatocyte hyperplasia. The hypertrophy may show a lobular distribution, with the pattern of lobular zonation and severity reflecting species, strain, and gender differences in addition to effects from specific xenobiotics. Toxicity and hepatocarcinogenicity may occur when liver responses exceed adaptive changes or induced enzymes generate toxic metabolites (Table 5.5). These undesirable consequences are influenced by the type and dose of xenobiotics, and show considerable species differences in susceptibility and severity that need to be understood for assessing the potential effects on human health from similar exposures to specific xenobiotics.
Hormones can also induce hypertrophy. For example, injection of growth hormone from the anterior pituitary induces hypertrophy of liver cells, which have an increase in their RNA content. Whether the various types of hypertrophy are considered physiological, adaptive, or pathological depends on the philosophy of the person making the judgment. For example, because hypertension is a disease, then cardiac hypertrophy that occurs secondary to the hypertension can be considered pathologic. However, one could argue that cardiac hypertrophy is an adaptive physiological response to an increased demand for work, regardless of the proximate cause.

1.6. Metaplasia and Anaplasia (Atypia)
Qualitative changes such as metaplasia and anaplasia (or atypia) can occur in hyperplastic cells and represent one of the hallmarks of pathological hyperplasia. However, qualitative changes in the phenotype or in the association and organization of groups of cells can also occur in the absence of clear-cut evidence of increases in cell number.
Metaplasia is the reversible substitution of one type of fully differentiated cell for another within a given tissue, and is seen most commonly in epithelial tissues. The notion that metaplasia usually involves the substitution of a less specialized cell type should be avoided in that it involves a value judgment regarding whether a cell expressing a different set of genes is indeed less specialized. For example, squamous metaplasia occurring in an area normally populated by ciliated respiratory epithelium represents a situation where fully specialized squamous epithelium provides protection against irritation. Removal of the irritating stimulus results in replacement by fully specialized ciliated epithelium.
1.7. Benign vs malignant neoplasms
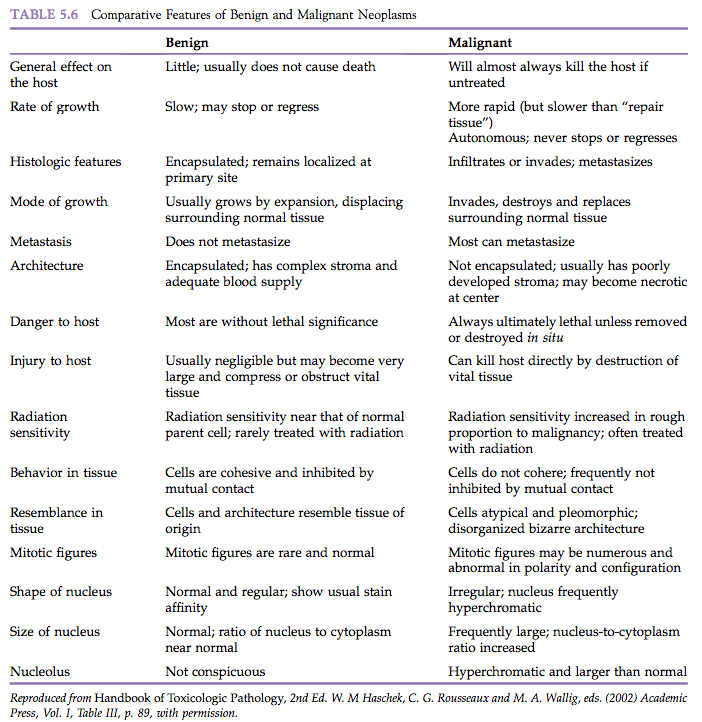
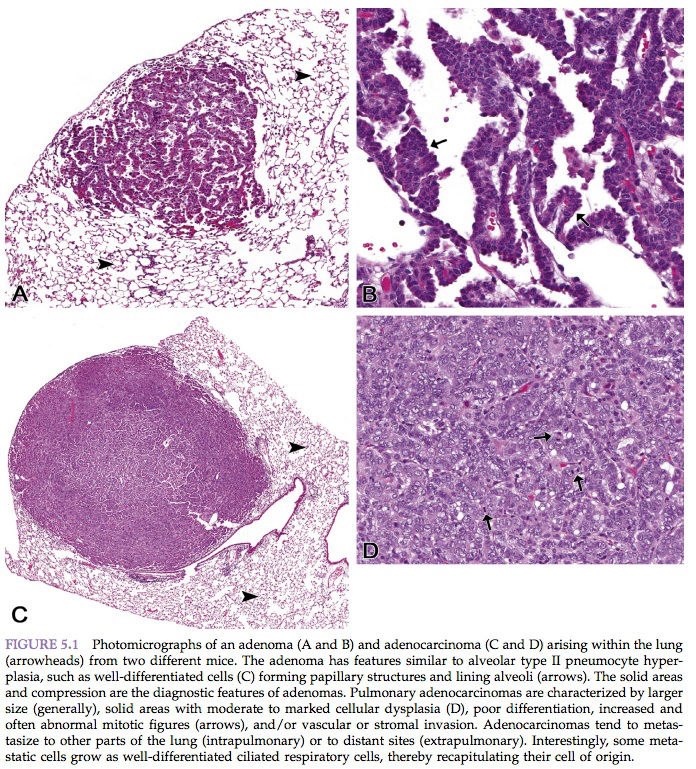
“Benign” comes from the Latin word benignus, and means innocuous. A benign neoplasm is a localized growth of well-differentiated, noninvasive tissue (Figure 5.1). Its growth is by expansion, and it may produce compression of adjacent normal tissues. Benign neoplasms ordinarily grow very slowly and are usually not life-threatening unless they interfere with vital functions, such as in a cardiac Schwannoma or a so-called “benign” meningioma severely compressing vital areas in the brain or spinal cord. Characteristic features of benign neoplasia are listed in Table 5.6.
Controversy regarding the significance of benign neoplasia with respect to the development of malignancy is similar to that associated with preneoplastic lesions. In chemical carcinogenicity tests using rodents, carcinogens frequently produce both benign and malignant neoplasms of a given tissue, and morphologic evidence exists that the benign lesions progress to malignancy in some studies.
“Malignant” comes from the Latin word malignus, and means malicious. Malignant neoplasms grow rapidly and are characterized by local invasiveness. Areas of necrosis seen in some malignant neoplasms presumably result when growth is so rapid that the neoplastic tissue outgrows the existing blood supply. Malignant growth is disorganized, and such neoplasms may spread by extension into adjacent tissues or by metastasis to distant sites via blood and/or lymphatic circulation (Figure 5.1). Characteristics of malignant neoplasms are listed in Table 5.6.
Some malignant neoplasms, particularly carcinomas, at some time in their evolution are at an intraepithelial or in situ stage. In situ carcinomas are microscopic lesions that have cytological criteria of malignancy, but are localized and have not gone beyond the basement membrane (Table 5.6).
2. CANCER IS A GENETIC DISEASE
2.1. Overview
The carcinogenic process involves the alterations of four broad categories of cancer genes, namely the activation of oncogenes, inactivation of tumor suppressors, evasion of apoptosis genes, and defective DNA repair genes (Table 5.7). Of the approximately 20 000 genes in the mammalian genome, hundreds are known or proposed oncogenes while relatively fewer are tumor suppressors, apoptosis evaders, or repair genes.

There are other fundamental cellular alterations, including limitless replicative potential, sustained angiogenesis, and ability to invade and metastasize. During the multistep process each neoplasm, arising from an individual cell, accumulates at least 80 genetic alterations in cancer genes, a dozen of which are considered “driver” mutations that drive the cancer’s uncontrolled growth. Multistep models of carcinogenesis have proven useful for defining events in the neoplastic process and form the cornerstone of current hypotheses of the biological mechanisms of carcinogenesis. Multistage models of carcinogenic processes have been demonstrated in a variety of organ systems, such as skin, liver, urinary bladder, lung, kidney, intestine, mammary gland, and pancreas, by utilizing various experimental animal models.
From use of these model systems, various agents have been categorized as initiators, promoters, and complete carcinogens. The operationally defined phases of carcinogenesis – initiation, promotion, and progression – are useful for discussion and understanding of carcinogenesis, but in fact each of these phases in the process may consist of multiple stages.
A large body of experimental data supports the contention that malignant cancer is generally irreversible. Even in initiation–promotion studies, the initial mutational changes constituting initiation may remain latent for weeks or months before being expressed by administration of a promoting agent. This “memory effect” is compatible with a stable somatic mutation.
Since the mid-1980s, oncogenes have been identified in the tumor DNA of many human neoplasms as well as in spontaneous and chemically-induced neoplasms in animals (Table 5.8). These oncogenes are frequently in the ras oncogene family, and the activated ras oncogenes frequently differ from their homologous proto-oncogene by virtue of a single point mutation.
In some experimental situations, carcinogen-induced activation of some oncogenes appears to be an early event in the carcinogenic process. While it can be argued that activation of proto-oncogenes may be a necessary event in the genesis of some cancers, it is unlikely that a single point mutation associated with an activated oncogene is sufficient in and of itself for the development of all cancers.

2.2. Oncogenes, Tumor Suppressors, Apoptosis and Repair Genes
Oncogenes are dominant-acting structural genes that encode for protein products capable of transforming the phenotype of a cell. Oncogenes were first identified as the transforming genes of retroviruses. These oncogenes were not necessary for the life cycle of the virus but were responsible for transforming virally infected cells and producing cancer in the host.
It was later learned that the transforming oncogenes were not intrinsic viral genes but rather were normal cellular structural genes captured from eukaryotic organisms previously infected by a retrovirus. The virus integrates in to the host genome and after rearrangement a portion of the normal cellular genes assimilates in to the viral genome, a process called transduction. The transduced oncogene in the viral genome is referred to as a viral oncogene (v-onc). The homologous gene in the host genome is called a cellular oncogene (c-onc) or a proto-oncogene.
In capturing the cellular proto-oncogene, the virus lost some of its own structural genes and consequently often became incapable of replicating in the absence of helper viruses. As the genomes of various mammalian and sub-mammalian species were examined, it was found that homologs to the retroviral oncogenes were present in species as diverse as yeast, fruit flies, amphibians, birds, and mammals. The high degree of evolutionary conservation of these proto-oncogenes suggested that they served important normal functions in the cell.
It is now known that proto-oncogenes encode for proteins that are important in cell growth, development, and differentiation. Because cancer is a perturbation of normal cell growth and differentiation, the potential significance of alterations in proto-oncogenes became apparent. It is thus not surprising that examination of DNA from human and animal tumors has demonstrated the existence of dominant transforming oncogenes, some of which correspond to those responsible for the carcinogenicity of acute transforming retroviruses.
An activated proto-oncogene has come to be called an oncogene, and is a proto-oncogene that has been altered quantitatively or qualitatively, resulting in inappropriate or overexpression. Activation can occur in several ways. Retroviral transduction had been shown to result in the acquisition of point mutations, deletions, or gene fusions within the coding sequence of the transduced proto-oncogene. This leads to abnormal functioning and changes in levels and schedules of expression of encoded protein products.
Retroviruses can also affect the expression of proto-oncogenes by a process called insertional mutagenesis. In this situation, the retroviral DNA integrates into the host-cell DNA adjacent to or within the coding sequence of a proto-oncogene. The powerful retroviral promoters (regulatory genes) then drive transcription of the normal or truncated gene product of the proto-oncogene. Activation of proto-oncogenes can also occur by mechanisms independent of retroviral involvement. Point mutations and DNA rearrangements, such as translocations or gene amplifications, can result in proto-oncogene activation by leading to altered levels or schedules of expression of the normal protein product, or in normal or altered levels of expression of abnormal protein.
The activation of proto-oncogenes in spontaneous and chemically-induced neoplasia has received considerable attention over the years. A variety of activated oncogenes have been documented in rodent neoplasms. From some experimental studies it appears that certain types of oncogenes are activated by carcinogen treatment, and that this activation is sometimes an early event in tumor induction. Other studies with human and rodent neoplasms suggest that oncogene activation is involved later in the carcinogenic process, specifically during tumor progression.
In vitro neoplastic transformation and somatic cell fusion studies provide evidence that malignant transformation represents a balance between genes for expression and for suppression of malignancy. There is also convincing evidence that growth suppressor genes, generally referred to as tumor suppressor genes, play a critical role in in vivo carcinogenesis. Growth suppressor genes are regulatory genes that normally function to limit or suppress normal growth by inhibiting the activity of structural genes responsible for growth. As such, when intact, they have a function opposite to that of oncogenes and might effectively oppose the action of an oncogene. While proto-oncogenes have to be activated to influence carcinogenesis, suppressor genes have to be inactivated for the transformed phenotype to be expressed. Inactivation can be achieved by chromosome loss, gene deletion, recombination, gene conversion, or point mutation.
Proto-oncogene activation is generally the result of a somatic mutation. Mutant forms of tumor suppressor genes might be present in germ cells and may thus be hereditary. It is generally necessary for both alleles of a tumor suppressor gene to be inactivated to ultimately result in cancer. Exceptions include inactivation of a normal allele by genomic imprinting or by dominant-negative mechanisms, as is proposed for the p53 tumor suppressor gene.
In common human cancers, multiple tumor suppressor genes may be affected, supporting the notion that cancer development involves perturbation of several levels of growth control. Inactivation of the tumor suppressor gene p53 is a frequent occurrence in a variety of human cancers. Thus, inactivation or loss of tumor suppressor genes, working in concert with the activation of oncogenes and with a variety of endogenous and exogenous stimuli, plays an important part in the complex process of carcinogenesis.
The pivotal role of cell proliferation in all phases (e.g., initiation, promotion, progression) of the multistep process of carcinogenesis is inextricably linked to positive and negative cell cycle control mechanisms as influenced by oncogenes, tumor suppressor genes, growth factors and their cognate receptors, hormones and their receptors, and the action of exogenous agents (e.g., chemicals and viruses) on cell cycle control. Uncontrolled cellular proliferation is the hallmark of neoplasia, and many cancer cells demonstrate damage to genes that regulate their cell cycles directly.
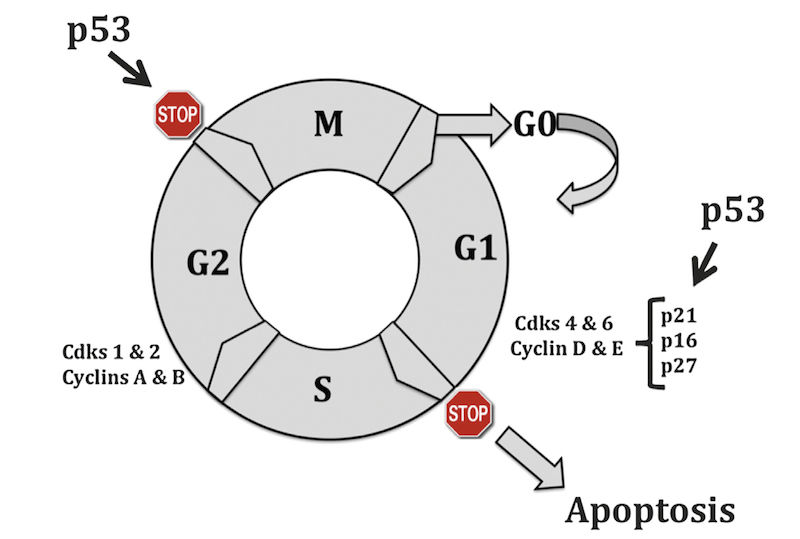
The prevailing model of the cell cycle is that of a series of transitions at which certain criteria must be met before the cell proceeds to the next phase. The cell cycle is composed of an S (DNA synthesis) and an M (mitotic) phase, separated by two gap phases (G1 and G2). Progression through the cell cycle is tightly controlled by a group of heterodimeric protein kinases comprising a cyclin as a regulatory element and a catalytic subunit known as a cyclin-dependent kinase (Cdk). There are many combinations of cyclin/Cdk complexes, and each phase of the cycle is characterized by a specific pattern of expression and activity.
Five major classes of mammalian cyclins (termed A–E) have been described. Cyclins C, D1–3, and E reach their peak of synthesis and activity during the G1 phase and regulate the transition from G1 to S phase. However, cyclins A and B1–2 achieve their maximal levels later in the cycle, during the S and G2 phases, and are regarded as regulators of the transition to mitosis. Association with cyclins not only activates cyclin-dependent kinases but also determines their substrate specificity. Depending on the cyclin partner and therefore the cell cycle stage, different key target molecules are phosphorylated. These events occur in a highly regulated temporal sequence that is maintained through a series of checkpoints (Figure 5.2).
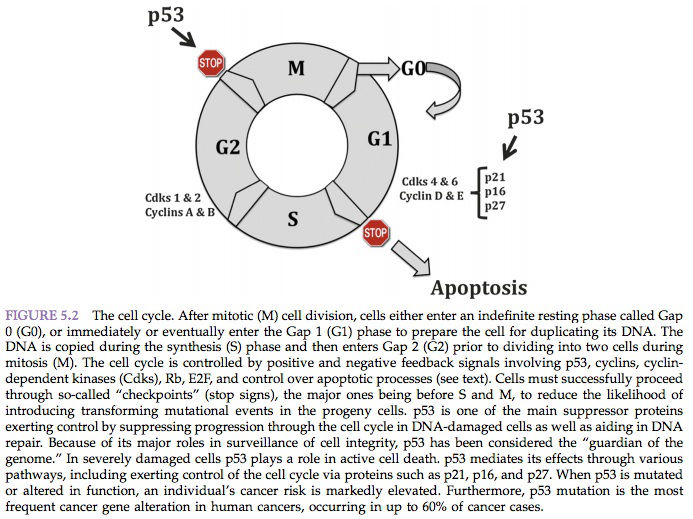
The presence of these checkpoints allows DNA repair before further progression into the cycle. The components of checkpoint control may not necessarily be essential to the workings of the cycle; instead, their role is to “brake” the cycle in the face of stress or damage. Abrogation of cell cycle checkpoints with agents such as methylxanthine analogs or pentoxifylline increases the cytotoxicity of DNA-damaging agents.
The importance of DNA damage in triggering a cell cycle shutdown is obvious. Replication of a damaged template would certainly result in irreversible chromosomal aberrations and a high mutation rate. Two major checkpoints are thought to be particularly important following DNA damage and have been established at the middle to end of G1 (preceding DNA replication) and G2 (preceding chromosome segregation).
Loss of the G1 checkpoint triggers genomic instability at the time of interaction of unrepaired DNA with the DNA replication machinery, leading to deletion-type mutations and aberrant gene amplification. Inactivation of the G1 checkpoint serves as an initiation step that makes the cell susceptible to unregulated growth (initiation), increasing the probability of subsequent genetic alterations and establishing the fully developed neoplastic phenotype. Control at the G1 checkpoint is dependent on cyclin D1 (degraded at the G1/S transition) and cyclin E (degraded in mid-S phase). Overexpression of either cyclin D1 or cyclin E and subsequent activation of the cyclin D1 and cyclin E/Cdk complexes results in entry into S phase and decreased G1 time.
Cyclin D1 is overexpressed in many human cancers, including breast and non-small cell lung carcinomas, sarcomas, melanomas, B cell lymphomas, and squamous cell carcinomas of the head and neck. Cyclin D1/Cdk4 complexes act to phosphorylate pRB, the product of the retinoblastoma susceptibility gene. pRB does not seem to possess sequence-specific binding activity, but instead exerts a negative regulatory effect on gene expression through complex formation with DNA-binding proteins, including members of the E2F family. In non-dividing or G0 (arrested) cells, underphosphorylated pRB is bound to E2F family members, leading to repression of E2F-mediated transcription.
Upon phosphorylation by cyclin/Cdk complexes, pRB dissociates from E2F proteins, leading to transcription of genes promoting S phase entry. Thus, underphosphorylated pRB maintains cells in G1, whereas phosphorylation inactivates pRB and allows exit from G1. In humans, inactivation of pRB is observed most commonly in retinoblastomas, osteosarcomas, carcinoid tumors, and non-small cell lung cancers.
Another tumor suppressor gene, p53, is necessary for G1 phase arrest after DNA damage. Mutations at the p53 locus are the most frequent genetic alterations associated with cancer in humans. The p53 protein can be divided into three main regions: the amino-terminal transactivation domain, the sequence-specific DNAbinding central core, and the multifunctional carboxy-terminal domain, which includes tetramerization and nuclear localization domains.
The majority of p53 mutations involve several highly conserved regions within the DNA binding core. Lack of p53 permits synthesis of damaged DNA and increases the incidence of selected types of mutations. This increased incidence has been shown after a variety of DNA damage mechanisms, such as ionizing radiation (strand breaks), alkylation by methyl-methane sulfonate (MMS), ultraviolet irradiation (photodimers), and a variety of environmental carcinogens. Thus, one of the major roles of p53 is to ensure that, in response to genotoxic damage, cells arrest in G1 and attempt to repair their DNA before it is replicated.
The wild-type p53 protein is normally kept at very low steady-state cellular levels by its relatively short half-life. However, it is stabilized and accumulates in cells undergoing DNA damage or in those responding to certain forms of stress. After DNA damage, p53 binds a consensus-binding site and activates the transcription of several “downstream” genes.
One of these genes codes for the p21 protein. The p21 gene belongs to a family of negative cell cycle regulators, which function as cyclin-dependent kinase inhibitory molecules. Genes that encode these proteins are designated CKI genes. These negative regulators form stable complexes with cyclin/Cdk units and inactivate them. p21 inactivates cyclin E-Cdk2, cyclin A-Cdk2, and cyclins D1-, D2-, and D3-Cdk4 complexes, thereby inhibiting pRB phosphorylation and preventing progression of the cell cycle beyond G1.
The p53 protein also activates the BAX gene, involved in the regulation of apoptosis. Apoptosis is a cell suicide mechanism that leads to “programmed cell death.” Apoptotic cells undergo cell shrinkage and chromosomal condensation in response to DNA damage. These changes prevent the replication of cells that have sustained a degree of genetic damage beyond repair. The p53 protein regulates its own function through the activation of the MDM2 gene. The product encoded by MDM2 is a 90-kDa zinc finger protein (mdm2), which also contains a p53-binding site. The mdm2 protein binds to p53 and acts as a negative regulator, inhibiting wild-type p53 transcriptional activity and creating an autoregulatory feedback loop.
A pRB-binding site has also been identified at the carboxy-terminal domain of mdm2 that interacts with pRB and restrains its functions. Thus, overexpression of mdm2 inactivates both p53 and pRB in a fashion similar to some viral oncoprotein products, demonstrating a potential link between p53 and pRB in cell cycle regulation, apoptosis, and tumor progression.
The cell cycle and its regulatory proteins are altered to the benefit of many viral agents. Progression of the cell cycle is advantageous for many viruses, as they require activation of host replication machinery to replicate their own genome. Several viral proteins have been shown to interact with and alter p53 and pRB.
The SV 40 large T antigen, the adenovirus EIB 55-kDa protein, and the human papillomavirus E6 protein each bind to p53 and inactivate it. Similarly, complex formation of underphosphorylated pRB with the SV 40 large T antigen, the adenovirus EIA protein, and the papillomavirus E7 protein leads to pRB inactivation and cellular immortalization. High-transformation strains of tumor viruses encode proteins that bind and inactivate both p53 and pRB. In the normal cell, the growth deregulation caused by pRB inhibition can be counteracted by apoptotic cell death produced by normal p53.
With the loss of both p53 and pRB, E2F activation stimulates unchecked cellular proliferation, leading to the emergence of neoplastic cell growth. The high rate and mutation pattern of p53 and pRB in primary tumors have rendered them prototype tumor suppressor genes. Furthermore, detection of p53 and pRB mutations and altered expression of their encoded products appear to be of clinical prognostic significance when identified in specific cancers.
Additional gene products activated in response to DNA damage include transcription factors, growth factors, growth factor receptors, protective enzymes, and proteins associated with inflammation and tissue injury and repair. These findings are consistent with the current understanding of the molecular basis of carcinogenesis as a multistep process. Therefore, it is essential to further advance our understanding of the intricate molecular mechanisms that govern chemical carcinogenesis. This will allow us to improve strategies for assessing human cancer risk and to design effective treatment regimens.
2.3. Cell Proliferation and Apoptosis
Increased cell proliferation can make an important contribution to the process of carcinogenesis. This increase can come about in either or both of two ways. First, the enhanced cell turnover could lead to “fixation” of spurious or spontaneous genotoxic damage. DNA damage is believed to occur continually from cellular exposure to endogenous or exogenous genotoxic insults or simply from endogenous errors in DNA replication. Fortunately, the vast majority of such DNA damage is repaired prior to cell division by efficient cellular enzymatic systems. The faster the cells are dividing, the greater the chance that genotoxic damage would not be repaired prior to cell division. Once cell division has taken place, the genomic alteration is “fixed” and is hereditary. If such a genomic alteration confers a selective growth advantage to the cell and its progeny, there will be clonal expansion and progressive development of neoplasia.
A second way in which enhanced mitogenesis could contribute to neoplasia is to stimulate cell division in an already initiated cell, thereby providing the growth stimulus whereby it can expand clonally. In situations of continued exposure to a non-genotoxic mitogen, it is likely that both potential mechanisms could act in a complementary fashion. In either case, the mechanism of cancer “induction” would be considered secondary. A variety of situations exist in animal carcinogenicity models where non-genotoxic chemicals given at sufficiently high doses may play a causative role in the development of neoplasia simply by enhancing cell proliferation. Examples of agents that may operate through this potential mechanism are given in Table 5.9.

Confirmation of whether enhanced cell turnover is a realistic explanation for the observed carcinogenicity of non-genotoxic carcinogens will require carefully conducted studies that demonstrate enhanced cell turnover at doses that result in cancer, and no increase in cell turnover at non-carcinogenic doses. If enhanced neoplasia in an animal test system is a consequence of increased cell proliferation and if the increased cell proliferation is related to exposure to excessive amounts of certain chemicals, then it becomes important to place the animal test results into the context of expected environmental exposure in assessing potential risk to human health.
Current data strongly suggest that cell death may be as essential as cell proliferation in carcinogenesis. The ratio between cell birth and counterbalancing cell death determines tumor growth.
Two forms of cell death may be seen in cancer development: necrosis and apoptosis. Necrosis typically occurs when a developing cancer outgrows its blood supply. Apoptosis is an energy-dependent process that involves active gene transcription and translation. In preneoplastic lesions, apoptosis is the predominant form of cell death that is observed. Chemicals, food deprivation, certain cytokines, growth factors, tumor suppressor genes, and withdrawal of mitogenic agents in experimental in vivo carcinogenesis may trigger enhanced apoptosis. It has been reported that the growth of dioxin-promoted preneoplastic liver foci in rat hepatocarcinogenesis is due to inhibition of apoptosis rather than to enhanced cell proliferation. An underlying principle of cancer chemotherapy is the selective induction of apoptosis in neoplastic cells.
2.4. Somatic Mutation Theory
There is strong evidence that a critical step in carcinogenesis is a structural alteration occurring in the genetic machinery of a somatic cell. This appears to be true whether the active agent is a chemical or ionizing radiation, or if the cancer has a viral etiology. According to the mutational hypothesis, one or more point mutations are responsible for initial and/or critical steps in the neoplastic process. In 1914, the somatic mutation theory for cancer was put forward.
According to the somatic mutation theory, cancer originates when an otherwise ordinary cell undergoes a mutation. In 1953, it was postulated that if a large enough population of somatic cells lives for a sufficient length of time, gene mutations will occur in some of them. As the mutated cells proliferate, there is a finite probability that some of them will sustain at least a second mutation (Figure 5.3).
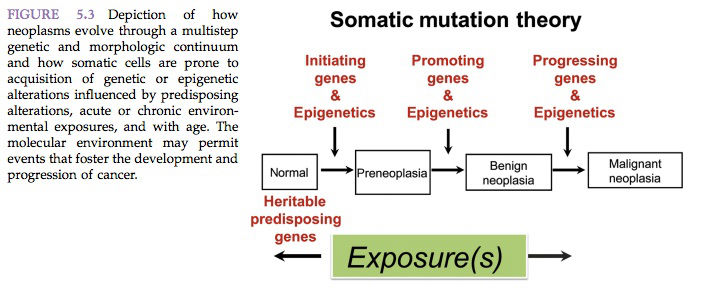
As the process of successive mutation and proliferation continues, cells eventually sustain enough genetic alteration to become autonomous and result in cancer. The accumulation of successive mutations would be expected to increase as a function of age and of the degree of cell proliferation. Early occurrence of cancer might be expected to result from exposure to mutagens as well as to agents that increase the rate of cell proliferation. The relevance of the somatic mutation theory to multistep carcinogenesis has been shown repeatedly to be the most probable explanation for the initiation of carcinogenesis (Figure 5.3).
In more recent times, the possibility that malignant cancer can result from multiple genetic insults has been championed by several investigators and is gaining widespread acceptance. There seems little doubt that genetic mutation plays a causative role in the genesis of some cancers. However, it has not been demonstrated unequivocally that mutation is a universal, sufficient, or necessary prerequisite for all cancers.
It has been proposed that neoplastic cells likely have a higher mutation rate than normal cells and thereby increase the likelihood of neoplastic cells acquiring further mutations conducive to neoplastic growth features. This was referred to as the “mutator phenotype.” It suggests that early mutation in stability genes (i.e. DNA repair, mismatch repair, DNA replication, or chromosome maintenance) will lead to the mutator phenotype and further mutations contribute to the subsequent invasive and metastatic properties of the neoplastic growth.
Numerical and/or structural chromosomal abnormalities and alterations are virtually universally present in neoplasms, particularly malignant neoplasms. Chromosomal aberrations are found in apparently spontaneous neoplasms as well as in those induced by chemical carcinogens or by oncogenic viruses. Against a backdrop of apparently random chromosomal aberrations observed in neoplasms, it is also apparent that there are a few constant karyotypic changes associated with specific neoplasms. Most notable is the association of the Philadelphia chromosome with the lymphoma/leukemia group of human diseases. However, in light of the relatively few constant karyotypic changes associated with specific histogenic types of neoplasms and the random nature of the preponderance of chromosomal aberrations observed in neoplasms, it is difficult to establish a cause–effect relationship between specific chromosomal aberrations and cancer. Non-random chromosomal aberrations in animal neoplasms include trisomy 15 in T cell lymphoma of the mouse, trisomy 13 in mouse mammary adenocarcinoma, and trisomy 4 in ethylnitrosurea-induced neurogenic neoplasms in the rat.
Reciprocal translocation is a form of gene rearrangement where portions of two chromosomes are simply exchanged with no net loss of genetic information. This can result in an alteration of the structure of the genes by virtue of their new location and/or in abnormal expression of the translocated gene(s). The Philadelphia chromosome associated with human lymphoma/leukemia represents a prototypical example of this phenomenon. This specific chromosomal abnormality consists of a translocation between the long arms of chromosomes 9 and 22, and is seen in 85% of patients with chronic myelogenous leukemia. Translocations between other chromosomes are associated with different forms of leukemia.
Gene amplification represents a situation where there is an increase in the amount of DNA present in a specific region of a chromosome. Chromosomal abnormalities observed in karyotype preparations such as homogeneously stained regions, abnormal banding patterns, and double minutes are the result of gene amplification. Assuming that the amplified genes are transcriptionally active, an excess of the product encoded by the amplified genes would be anticipated. Drug-induced gene amplification, possibly with an associated specific karyotypic change, is known to result in drug resistance due to an increase in the amount of gene product. A number of observations have been made with respect to gene amplification and oncogenesis. The classical skin tumor promoter, phorbol ester, causes amplification of specific genes in cultured cells. DNA amplification has been shown to occur at the site of insertion of oncogenic retroviruses.
It has been shown that increased production of normal protein products of proto-oncogenes (e.g., c-ras) that have been amplified may contribute to the malignant phenotype. Amplification of c-myc is seen in small cell cancer of the lung in humans, and the degree of amplification is correlated with clinical aggressiveness of this cancer. The causative role of DNA transposition, other types of DNA rearrangement, and even amplification of endogenous genes in either the origin or the development of neoplasia is difficult to ascertain at present. Even the mechanisms causing such somatic chromosomal abnormalities remain obscure.
However, the sites for chromosomal rearrangement seem to correspond to specific fragile locations in the genome that may be uniquely susceptible to breakage. Further documentation of chromosomal abnormalities in various neoplasms may provide important clues about basic genetic mechanisms associated with carcinogenesis. In some cases, specific chromosomal abnormalities have been shown to have prognostic significance. Also, modulation of gene products may foster malignant behavior by causing uncontrolled cell proliferation and metastasis.
2.5. Gene Regulation
MicroRNAs
MicroRNAs (or MiRNAs) are small noncoding, single-stranded RNAs, approximately 20 nucleotides in length, that control gene expression. They can incorporate into what is referred to as the RNA-induced silencing complex. Through the action of the RNA-induced silencing complex they “silence” gene expression via a post-transcriptional event. MiRNAs act to control various pathways, including cell growth, differentiation, and survival. MiRNAs have been shown to undergo changes in expression in cancer cells, and frequent amplifications and deletions of miRNA loci have been identified in many cancers. They act by increasing the expression of oncogenes or by reducing the expression of tumor suppressor genes.
MiRNAs can act like a tumor suppressor when the miRNA enhances expression of oncogenes that lead to overproduction of the oncogene product. Conversely, miRNA can reduce the tumor suppressor proteins’ expression and thereby act as an oncogene. MiRNA profiling of several human tumors has led to the downregulation or deletion of certain miRNAs in leukemias and lymphomas, resulting in increased expression of BCL2, the anti-apoptotic protein. MiRNA-mediated up regulation of ras and myc oncogenes has been detected in some lung tumors and B cell leukemias, respectively. Overexpression of specific miRNAs has been documented in a number of cancers in some human brain and breast tumors, but a critical oncogene or tumor suppressor gene has yet to be identified. Look for more about miRNAs and role in cancer in the coming years of exciting new understanding.
Epigenetics
Epigenetics refers to reversible, heritable changes in gene expression that occur without mutation. Such changes involve post-translational modifications of histones and DNA methylation, both of which affect gene expression. In normal, differentiated cells, the majority of the genome is not expressed. Some portions of the genome are silenced by DNA methylation and histone modifications that lead to the compaction of DNA into heterochromatin. On the other hand, cancer cells are characterized by a global DNA hypomethylation and selective promoterlocalized hypermethylation. Indeed, it has become evident during the past few years that tumor suppressor genes are sometimes silenced by hypermethylation of promoter sequences rather than mutation.
The non-mutational genetic hypothesis postulates that a genomic perturbation other than a mutation may lead to cancer. One of the mechanistic explanations for this hypothesis relates to changes in the pattern of DNA methylation. Altered patterns of DNA methylation are associated with exposure to chemicals and are tissue-specific. Consistent with a role of altered methylation in carcinogenesis are the observations that DNAs from specific tumors have evidence of hypomethylation; carcinogens are known to interfere with DNA methylation; and the drug 5-azacytidine, which causes under methylation of genes, has been shown to enhance cell transformation. In general, genes from cancer cells are hypomethylated as compared to their normal counterparts, although important qualifications that relate to the test system used should be noted.
Hypomethylation is believed to influence the regulation of transcription and gene expression and may be associated with cellular differentiation. Unfortunately, as with many abnormalities that are associated with cancer cells, we are faced with a dilemma in deciding if hypomethylation of genes is a cause of malignancy or a consequence of the altered metabolism of malignant cells.
Blocked Differentiation
An apparent characteristic of transformed cells is a partial or total block in terminal cell differentiation. A block of normal differentiation implies an alteration in normal gene expression. While it has been postulated that neoplasia may represent blocked or abnormal cellular differentiation, the paucity of knowledge relative to normal mechanisms that regulate differentiation makes it difficult to elucidate the potential role of abnormal cell differentiation in the genesis or development of cancer.
In fact, experiments exploring the possibility that neoplasia is a disease of blocked differentiation have shown that, under specific environmental conditions, malignant cells have an intrinsic ability to revert to a normal phenotype. This suggests that if blocked differentiation plays a role in the development of neoplasia, the endogenous and/or exogenous factors regulating differentiation must be present continually to maintain the neoplastic phenotype. It has been argued, however, that cancer is not blocked differentiation but rather represents a new state of differentiation conferring a selective growth advantage to the altered phenotype.
Intercellular Communication
Among the potential regulatory factors associated with the phenotypic expression of transformed cells are those associated with intercellular communication. Intercellular communication (sometimes referred to as metabolic cooperation, based on a popular assay for its measurement) is known to play an important role in the phenotypic expression of some transformed cells in vitro. In co-cultivation studies, the presence of normal cells is sufficient to prevent the phenotypic expression of transformed cells.
Apparently there is sufficient communication between cells, most probably occurring via gap junctions, such that biochemical substances elaborated by the normal cells block the expression of an abnormal phenotype by the transformed cells. Although not a ubiquitous characteristic of non-genotoxic carcinogens, it has been suggested that the inhibition of cellular communication may be the mode of action of many, but not all, chemicals that induce neoplasia but do not affect the genome directly.
Current studies have shown that many tumor-promoting agents inhibit gap junction intercellular communication associated with the aberrant expression of connexin and the loss of function of cell adhesion molecules. It has also been suggested that the inhibition of intercellular communication may be one unifying mechanism for all forms of neoplasm promotion. Non-cytotoxic levels of chemicals known to be promoters in lung, liver, skin, colon, breast, or esophagus have been shown to inhibit intercellular communication in cell culture systems.
An in vivo phenomenon that might be considered a different form of intercellular communication relates to the postulation that epidermal chalones are instrumental in the promotion of skin cancer. Chalones are tissue-specific substances that inhibit the proliferation of immature cells and probably operate through a cell membrane receptor mechanism rather than via gap junctions.
Chalones are believed to be produced by neighboring cells, and may operate as a type of paracrine control mechanism. Skin tumor promoters, such as phorbol esters, possibly inactivate the chalone receptor site, thereby switching off the mechanism of growth control in the affected cell(s). The existence of chalones has been debated. Some regard them as hormonelike substances with inhibitory activity. A number of growth-inhibitory substances have been described (e.g., TGF-13). Many of these do not meet the tissue-specific definition of a chalone but nonetheless appear to regulate normal but not tumor cell growth.
Hormones
Hormones are chemical messengers that bind to specific cellular receptors and form a hormone–receptor complex that triggers a cellular response (see Endocrine System, Chapter 58). The cellular response is specific for both the hormone and the target cell. The target-cell response to hormone stimulation is typically an increase or decrease in cell division, or an acceleration or deceleration in differentiation. There is an almost continual discovery of endogenous messenger substances and growth factors that fit the broad definition of “hormone.”
Like hormones, hormone-related peptides, commonly referred to as growth factors, play an important role in the control of growth and differentiation of cells, tissues, and organs. A major difference between hormones and growth factors is that hormones are produced in an endocrine tissue and act on cells at a distant site, whereas growth factors are secreted by a variety of normal and abnormal tissues and act on nearby responsive cells.
Endogenous hormones have long been known to be associated with the development of specific neoplasms and in some cases with the inhibition of carcinogenesis (e.g., diethylstilbestrol, DES). Hormones or hormone imbalances undoubtedly play a major causative role in cancers of certain hormone-sensitive tissues (ovary, uterus, prostate, testes, endocrine organs). However, it is unlikely that they play a major causative role in the development of neoplasia in non-hormone-sensitive tissues. Through the use of two-stage animal models for mammary and thyroid cancer, hormones have been demonstrated to function as tumor promoters of neoplasia. There is little evidence that there is direct interaction of endogenous hormones with DNA, although minimal DNA binding has been shown with some synthetic estrogens, and some hormones do bind to protein.
Most endogenous hormones are believed to act through a specific cellular receptor mechanism. Once there is binding to a specific membrane, cytosolic, or nuclear receptor, the receptor becomes activated and intracellular activity occurs that affects transcriptional or translational processes, resulting in the synthesis of specific proteins. Just how hormonal interaction with a cell receptor leads to cancer is unknown, but one potential mechanism relates to an increase in cell turnover among cells that already possess latent genetic change.
Thus, endogenous hormones may serve to promote spontaneously or otherwise initiated cells. Alternatively, hormonal imbalance could lead to increased proliferation in a sensitive cell population with secondary genotoxic damage from any one of several environmentally prevalent genotoxic agents. This certainly is consistent with the frequent observation of endocrine tumors developing against a previously existing background of hyperplasia. Furthermore, the two possibilities are not mutually exclusive.
The precise signals and mechanisms by which hormones and related growth factors regulate cell proliferation and differentiation remain to be identified, and are areas of active research. Evidence suggests that the mechanism of action of hormones and growth factors may be closely associated with and related to the action of oncogene protein products.
2.6. Cancer Stem Cell Theory
For more than 100 years cancers have been recognized as morphologically heterogeneous populations of cells that arise from a single cell, but it has become clear in the past decade that functional heterogeneity also exists. Now, many studies suggest that a distinct subpopulation of cancer cells designated as “cancer stem cells” (CSC) sustain cancer growth. Specifically, only a minority of tumor cells (CSCs) from some of these cancers have the capacity to regenerate a tumor and sustain its growth when injected into immune-compromised mouse.
There is extensive evidence that multiple types of cancers arise from transformation of stem and/or multipotent progenitor cells during carcinogenesis; therefore, it is reasonable to consider that cancer in some cases may be a primary disorder of stem cell differentiation. Cancer stem cells, as these transformed stem cells are defined, are neoplastic cells that have stem cell properties, including the ability to self-renew in order to propagate additional malignant stem cells, and differentiate to give rise to phenotypically diverse non-carcinogenic cells.
Given these characteristics, these transformed stem cells therefore have an indefinite ability to self-renew, which is the driving force behind carcinogenesis. This is the “cancer stem cell hypothesis.” This hypothesis is not new to the field of cancer research. The formal cancer stem cell hypothesis was first proposed over 150 years ago: that these dormant embryonic components exist in adult tissues and may be activated to later become tumorigenic. Over the decades, the idea that tissue-specific stem cells may be the cell of origin of various cancers and that cancers may represent a maturation arrest of stem cells advanced the study of the cancer stem cell hypothesis. Indeed, the cancer stem cell hypothesis continues today as an enormous focus of the field of cancer research.
Through the ability to self-renew, a small population of carcinogenic stem cells within a neoplasm would have the ability to maintain malignant clones. Self-renewal is a process by which a stem cell pool maintains its numbers through symmetric and asymmetric division. In symmetric cell division, the progeny are identical to the initial stem cell; in asymmetric stem cell self-renewal, one of the two progeny are identical to the initial stem cell, whereas the other cell is a committed progenitor cell, which undergoes cellular differentiation. The traditional clonal origin of carcinogenesis states that transformation occurs through a series of sequential mutations resulting in preneoplastic lesions that progress to neoplasia and, finally, metastasis.
However, in many tissue sites in which cancers arise, cell populations have a high rate of cell turnover and thus do not persist for significantly long enough time to accumulate the necessary number of mutations sufficient for cellular transformation. On the other hand, through the tightly regulated process of self-renewal, normal stem cells are able to function over the lifespan of the host, and thus their longevity and continued mitotic activity makes them a significant target, and potential reservoir for, the accumulation of the numerous genetic mutations needed for transformation.
Many similarities between normal stem cells and tumor cells exist, and tumorigenic cells display functional and phenotypic qualities of the normal cells from which they arise. There is significant evidence that neoplastic cells undergo self-renewal and differentiation similar to that of normal stem cells. Both cancer cells and stem cells have indefinite proliferative potential, as well as the ability to undergo organogenesis and give rise to new tissues, although disorganized and atypical in the case of tumorigenesis. Both cancerous and normal tissue is composed of heterogeneous cell populations with various morphologies and proliferative ability.
Stem cells and various neoplastic cells express telomerase, and have anti-apoptotic mechanisms, and increased membrane transporter activity (thus being able to efflux chemicals and chemotherapeutics from the intracellular compartment). Telomerase is a type of reverse transcriptase that preserves the length of telomeres. It is active during cell replication, and plays a role in proliferation and immortality of cancer cells. The telomeres of chromosomes of normal somatic cells shorten during each cell division and are a part of cell senescence. In cancer cells, telomerase may be expressed and lengthen telomeres, contributing to cell immortality and autonomous neoplastic growth. Both stem cells and neoplastic cells have the ability to migrate (or rather metastasize in the case of neoplastic cells), and both share embryonic markers such as β-HCG and AFP, and several commonly recognized stem cell markers such as c-kit and CD34 are found in a variety of tumors.
The process of tumorigenesis in many types of cancer involves dysregulation of embryonic and developmental pathways involved in regulation of embryogenesis, cell fate determination, and stem cell maintenance, including Wnt, Notch, and Sonic Hedgehog pathways. In many ways, neoplastic cells differ from stem cells only in terms of their unrestricted and disordered growth and genetic alterations.
Significant evidence exists for the existence of tissue-specific cancer stem cells in various organs, consistent with the theory that cancers arising in various tissues originate from transformation of tissue-specific stem or progenitor cells. Identification of these cells can be problematic, because stem cells represent a very small proportion of the overall cell population in an organ.
In order to identify cancer stem cells, investigators have used a stem cell immunophenotype based on immunocytologic and immunohistochemical expression of various stem cell markers and cell surface markers, followed by cell sorting to identify a cancer stem cell population. Stem cell populations have been identified using panels of a number of immunocytochemical markers, including CD133, CD34, CD38, CD24, CD44, SOX2, OCT4, BMI1, ESA, Nanog, Nestin, and c-kit.
Significant biologic evidence for the role of stem cells as oncogenic targets of mutagenesis was initially reported in hematologic malignancies such as leukemia and multiple myeloma. In these diseases, it was documented that only a small subset of tumor cells was actually capable of extensively proliferating, and that the progression of the neoplastic disease was driven by this population that possessed stem or progenitor cell characteristics. In human acute myelogenous leukemia (AML), using the hematopoietic cell surface phenotype CD34+/CD38–/Thy1+, investigators found that the small population of AML cells possessing this phenotype could repopulate and induce leukemia in immunocompromised mice, with the same morphology and cell surface marker expression. Those cells that did not exhibit such a phenotype were unable to propagate, grow, and reproduce tumors in immunocompromised mice.
While the earliest evidence of cancer stem cells comes from studies in hematologic malignancies such as human acute and chronic leukemia and myeloma, multiple researchers have provided evidence for the existence of cancer stem cells in solid tumors, including brain, lung, colon, ovarian, pancreas and prostate tumors, as well as in breast cancer and melanoma, among others. Through molecular investigation, the use of stem cell markers, developmental pathways, and xenograft transplantation experiments in mouse models, numerous studies have provided significant evidence for the relevance of cancer stem cells as initiators of various types of cancer in humans.
A mouse xenograft model has been used to identify neural cancer stem cells through the use of a stem cell marker, CD133, and provided evidence that injection of as few as 100 of these cancer-initiating cells produced a tumor in mice that could be serially transplanted and was morphologically identical to the original neoplasm. Alternatively, injection of 105 CD133- negative cells failed to produce a tumor when transplanted into mice, indicating that only brain tumor cells possessing a stem cell phenotype were able to survive and proliferate to repopulate the neoplastic population, and that the non-stem cell tumor population failed to promote neural cancer. Identification of these brain tumor-initiating cells gives strong support for the cancer stem cell hypothesis as the basis for many solid tumors.
Studies on human lung cancer provided evidence that bronchioalveolar stem cells were putative stem cell targets of oncogenic KRAS, and when transformed were the precursors to lung tumors in vivo in mice. These stem cells normally maintain Clara cell populations in the bronchioles and alveolar epithelial cells within the lung. When transformed, these multipotent progenitor cells gave rise to pulmonary adenocarcinomas, contrary to previous belief that differentiated pulmonary alveolar or bronchiolar cells were the cell of origin for this type of cancer. It has been shown that, similar to brain tumors, tumorigenic cells in small cell and non-small cell lung cancer in humans are a rare population of CD133-positive, undifferentiated cancer stem cells that are able to self-renew, grow indefinitely, and repopulate secondary transplanted hosts, whereas CD133- negative tumor cells are not.
The CD133 cell surface marker has also been used to identify colon cancer-initiating cells using a similar xenograft model, in which only the small proportion of CD133+ colonic adenocarcinoma cells were able to maintain growth and recapitulate the original tumor morphologically in secondary hosts.
There is mounting evidence supporting a transformed stem cell in malignant melanoma. Melanomas are comprised of a heterogeneous cell population, and stem and progenitor cell markers have been detected in malignant melanoma cells in humans. Several studies have examined the presence of tumorigenic subpopulations of human malignant melanoma cancer stem cells expressing markers of stem cells (including CD133, CD166, and Nestin, CD20, and the ABCG2 transporter) associated with high tumorigenic potential, and found that melanoma cells expressing these markers had higher tumorigenic potential when transplanted into immunocompromised mice.
The existence of mammary epithelial stem cells was demonstrated over 40 years ago through the use of transplantation studies which showed that the entire repertoire of the gland, including fully functional mammary epithelial outgrowths containing ducts, lobules, and myoepithelial cells, could be generated from epithelial fragments taken from various regions of the mammary gland at different stages of postnatal development in the mouse.
In 1998, it was shown that multipotent stem cells present in the mature mouse mammary gland had the capability of self-renewal, and that mammary gland outgrowths in secondary recipients of transplants were derived from self-renewing stem cells. In 2006, researchers demonstrated that an entire functioning mammary gland could be recapitulated from injection of a single mammary stem cell possessing the immunophenotype Lin–/CD29high/ CD24+. Interestingly, using the stem cell phenotype (CD44þ/CD24, ESAþ), breast cancer stem cells reconstituted secondary xenograft hosts with as few as 200 tumor cells from primary human breast carcinomas, whereas tumor cells without this phenotype failed to produce tumors despite transplantation of thousands of cells.
The cancer stem cell origin hypothesis does not discount the somatic mutation theory, and it is possible that each paradigm could be contributing to transformation and ultimate formation of a neoplasm. The cancer stem cell hypothesis accepts the contribution of genetic instability, mutation, and epigenetic factors that influence the evolution of heterogeneity in a malignancy; however, in this case, the target of the genetic or epigenetic alterations is the stem cell, rather than the somatic cell.
Furthermore, the concept of initiation and promotion that is a significant part of the somatic mutation theory applies to the cancer stem cell hypothesis, in that mutations that lead to clonal expansion of a neoplasm may initially occur in a stem cell. In further support of stem cells as targets of transforming mutations and progression towards malignancy, there are several similarities between normal stem cells and tumor cells, and neoplastic cells indeed portray both functional and phenotypic aspects of the stem cells they originate from.
The idea that the growth of a solid tumor is driven by proliferation of cancer stem cells has profound implications for cancer therapy. Treatment of cancer as an homogeneous population of proliferative clones with unlimited growth potential is problematic, because many tumors may initially respond to therapy only to recur, often months to years later in life. If, in the view of the cancer stem cell hypothesis, solid tumors are considered to be composed predominantly of an heterogeneous population of non-proliferative tumor cells and a small subpopulation of transformed stem cells that predominantly drive carcinogenesis, this would account for the recurrence of tumors following an initial regression period, since this subpopulation of cancer stem cells is more resistant to current chemotherapeutics.
The current dogma is that cancer cells acquire resistance to chemotherapeutics as they evolve and undergo additional mutations; an alternative explanation to account for drug resistance is that the cancer stem cell population inherently possesses the ability to resist current therapies, and thus is able to contribute to disease progression or tumor reoccurrence.
A major cause of cancer-related death, metastasis, is currently considered to occur through sequential acquisition of mutations leading to selection of clones with metastatic potential; however, it has been shown for decades that in certain cancers a small number of individual cancer cells disseminate and lie dormant at distant sites in a state of quiescence for up to several years, until triggered to outbreak. Alternatively, some disseminated tumor cells never result in the formation of metastatic tumors. One well-accepted explanation is that such cells are destroyed by immune surveillance. Alternatively, one may consider that under the cancer stem cell hypothesis a tumor is composed largely of transformed but poorly proliferative tumor cells, and that only a small proportion, the highly carcinogenic cancer stem cell subset, is able to form a new tumor or metastasize.
If the growth and metastasis of tumors are driven by a small fraction of highly tumorigenic cancer stem cells within a heterogeneous population of tumor cells, this may explain the failure to develop therapies that are able to consistently eradicate solid tumors. Therefore, a paradigm shift in cancer therapy should be considered, in which therapy is directed towards killing cancer stem cells, which may result in much more effective therapies.
2.7. Tumor Regression
Regression of foci of altered hepatocytes and hepatocellular neoplasms has been occasionally described in rats and only rarely in mice. Mechanisms for tumor regression are unclear, but this phenomenon suggests that some histologically-appearing benign and malignant “neoplasms” depend on the sustained presence of the inciting chemical. In a study of chlordane there was evidence of regression of benign and malignant hepatocellular neoplasms after cessation of exposure in B6C3F1 mice. H-ras mutations, which commonly occur in up to 60% of neoplasms in this strain of mouse, were not present.
Other non-genotoxic murine hepatocarcinogens that induce tumors with a lower frequency of H-ras mutation than occurs in spontaneous tumors include hexachlorobenzene, Aroclor, phenobarbital, ciprofibrate, dieldrin, chloroform, and methyl clofenapate. The absence of ras mutations and evidence for regression implies that the reversible “neoplasms” may lack the appropriate genomic mutations necessary for autonomous proliferation. Alternatively, chlordane may provide a selective growth advantage to the hepatocellular lesions without ras mutations, eliminate ras mutated cells through hepatocyte necrosis or apoptosis, or induce tumorigenic pathways that do not involve ras activation. There was no histological evidence of immune-mediated rejection of any neoplasms in the study. Interestingly, bcl2 and bclx, proteins that inhibit apoptotic pathways, were elevated in hypertrophied centrilobular hepatocytes and liver neoplasms from rodents treated with chlordane, phenobarbital, or clofibrate. Additionally, a gene array profiling study supported the involvement of growth-promoting factors and oncogenes in chlordane hepatocarcinogenesis.
Complete regression of hepatocellular carcinomas occurs, rarely, in humans with cirrhosis and in children after cessation of androgen therapy. Benign liver tumors have regressed after withdrawal of oral contraceptives in women. Recent evidence suggests that chlordane can act by mimicking sex steroids, or by altering the synthesis and elimination of sex steroids or their receptors. Since some cases of tumor regression appear to be related to hormone dependency, such as the androgen- or contraceptive-induced liver tumors described in humans, perhaps chlordane acts by a similar mechanism.
Liver tumor regression has also been observed after cessation of chronic exposure to a variety of non-genotoxic rodent hepatocarcinogens, including chlordane, nafenopin, phenobarbital, and Wyeth 14,643 (a peroxisome proliferator) (Table 5.10). In contrast, some other non-genotoxic murine hepatocarcinogens, such as methylene chloride and dieldrin, induce hepatocellular neoplasms that persist and progress after cessation of prolonged exposures. Perhaps chlordane-induced neoplasms develop by a series of changes that progress from a dependent to an autonomous state. For example, some histologically benign and malignant liver neoplasms are sustained by the continual presence of chlordane, and with cessation of exposure only the autonomous neoplasms persist and some eventually metastasize. It also appears that pathways of chlordane-induced hepatocarcinogenesis progress independently of ras activation.
The findings suggest that chlordane appears to act through enzyme induction as well as non-genotoxic, hormone-like mechanisms that promote cell survival and inhibit cell death. If such mechanisms (Table 5.11) can be further elucidated, then we can more accurately assess the human risks of chemicals such as chlordane.

3. IDENTIFYING CARCINOGENS
3.1. Animal Bioassays
Widespread and routine evaluation of chemicals for their carcinogenic potential began in earnest in the mid-1960s with the use of a standardized protocol for the “bioassay” program of the National Cancer Institute (NCI). The original NCI “bioassay” has evolved to the present day National Toxicology Program (NTP) 2-year and perinatal carcinogenicity study.
Throughout this period a variety of alternative in vivo and in vitro assay schemes have been introduced in the hopes of identifying potential carcinogens in less time and for less money. Some of these alternative testing models have been highly beneficial tools to help explore the processes of carcinogenesis and the types of carcinogens, to identify the genotoxicity of chemicals, and to identify the specific mechanisms whereby given agents produce a carcinogenic response in rodents. Testing batteries, tier approaches, and decision-point analyses have been proposed to provide a basis for prioritizing chemicals in the testing queue and to provide a larger database for risk assessment.
These efforts serve to emphasize that, in the last analysis, there is no ideal test for identifying potential human carcinogens. In general, efforts to supplant the long-term carcinogen “bioassay” with less costly short-term tests have been frustrated by poor concordance of the latter with the traditional long-term test results. In the meantime, increasing costs for conducting long-term carcinogenicity studies have resulted in fewer chemicals being tested annually for potential carcinogenicity. Currently, much thought is being given to developing medium-term tests, such as 6- to 9-month studies in genetically modified mice, for carcinogenicity concomitantly with analyses of reasonable ways to reduce the cost of the standard long term rodent carcinogenicity test.
The current strategy for identifying the potential carcinogenicity of chemicals involves systemic (including dermal) exposure of male and female rats and mice to high doses and fractions thereof of chemicals over a 2-year period. At the end of 2 years, the carcinogenicity of the chemical being studied is assessed by measuring the excess in neoplasm production above background levels of incidence and multiplicity, documenting the occurrence of rare neoplasms with negligible background levels, or demonstrating a reduced latency in neoplasm development in exposed animals versus controls.
If an agent is observed to produce neoplasia in the experimental animals, the agent is regarded as a potential human carcinogen. In countries throughout the world, legal requirements mandate that all new chemical agents and drugs be tested in animal bioassays to determine if they cause cancer in the test animals. Additionally, since the mid-1960s in the United States, the National Cancer Institute (NCI) and currently the National Toxicology Program (NTP) have collectively conducted animal bioassays on more than 600 chemical agents to assess their potential to cause cancer.
Of those 600 agents, about half (290) are identified as rodent carcinogens. A number of them are considered known or suspected human carcinogens. Fifty-seven percent of the 290 NTP rodent carcinogens (Table 5.12) induced at least an increase in liver neoplasms (primarily in B6C3F1 mice), 22% an increase in lung tumors, and 14% in renal (primarily in F344/N rat), 13% in mammary gland, 13% in hematopoietic, 12% in forestomach, 10% in thyroid, and 9% in endothelial neoplasms.

The original NCI “bioassay” was intended as a screen for carcinogenicity in the rodent. It was generally not intended to be used for risk assessment, determining carcinogenic potency, identifying subtleties of chronic toxicity, determining the mechanism of observed carcinogenic responses, or establishing pathogenesis of lesions (see Carcinogenicity Assessment, Chapter 27). The implication behind the original NCI “bioassay” was that if a chemical was found to cause neoplasms in treated animals, further studies would need to be designed and conducted to specifically determine parameters such as the mechanism of cancer induction, the nature of the dose–response relationship, chemical metabolism, and target organ dosimetry. The more definitive testing of positive chemicals has generally not occurred.
However, in the absence of such additional relevant data, “bioassay” results have been used as a basis for human risk assessment and regulation. In an effort to consciously expand the original NCI “bioassay” and to make the test results more relevant for the interpretations being applied, considerable evolution has occurred in the design, conduct, and interpretation of long-term rodent carcinogenesis tests conducted under the auspices of the NTP.
While the maximum tolerated dose (MTD) concept has been retained in principle, it is applied more conservatively in setting the high dose. However, a fundamental difference still exists in how the NTP selects the high dose in a chronic rodent carcinogenicity study and how industrial organizations determine the high dose in their otherwise similar chronic rodent carcinogenicity studies. High-dose selection relates to philosophic approaches to testing for toxicity and carcinogenicity. Governmental and similar organizations have traditionally conducted their studies to determine if exposure to a chemical can be harmful under any circumstances, whereas industry most frequently conducts its tests to determine if the chemical is reasonably safe.
The first philosophic approach would tend to lead to situations where the animals might be overdosed, whereas the second approach would tend toward underdosing. There is no right or wrong to either philosophy, so long as the study results are interpreted appropriately. In addition to the high dose or EMTD, at least two additional lower doses are employed in NTP rodent carcinogenicity studies with a conscious effort to utilize human and environmental-exposure information to set the low doses. This permits better definition of dose–response and allows better utilization of data for risk assessment (see Risk Assessment, Chapter 31).
To fully characterize the toxicity of chemicals, modifications to the chronic rodent carcinogenicity study protocol have been selectively introduced by the NTP. Interim sacrifices and “stop studies” are incorporated into the design of many carcinogenicity studies to better define the pathogenesis and biological relevance of the anticipated response. The route of administration of chemicals is chosen to mimic natural routes of human exposure whenever possible. Consequently, corn-oil gavage of chemicals is rarely used in contemporary study design. Ancillary studies, such as chemical disposition and metabolism, toxicokinetics, reproductive toxicity, teratology, behavioral testing, and immunotoxicity, frequently complement contemporary 2-year carcinogenicity studies. In addition, in vitro and in vivo genotoxicity tests are conducted for each chemical.
The NTP and others who assess carcinogenic activity of environmental or pharmaceutical agents have utilized a number of animal models. The majority of NTP studies have been conducted with the Fischer344/N (F344) rat and the B6C3F1/N mouse (a hybrid of the C57BL/6 and C3H parental strains). The F344 rat is prone to developing nephropathy, mononuclear cell leukemia, seizures, and/or testicular interstitial (Leydig) cell tumors, whereas the B6C3F1 mouse is genetically susceptible to the spontaneous development of liver and lung cancer. These spontaneous lesions can sometimes interfere with the interpretation of the bioassay. More recently, the NTP has examined the utility of the Wistar Han and the Sprague-Dawley rat to replace the F344 rat, and has completed analysis of genetically modified mice – p53 deficient, Tg.AC v-HA-ras transgenic, and p16(INK4a)/ p19(ARF) – for screening carcinogens.
Additional carcinogenicity models have been developed, including a rat urinary bladder model, a rat pancreatic cancer model, a gastric cancer model, a rat thyroid cancer model, a fish liver neoplasm model, and rat and mouse colon cancer models. Each has its own limitations. A short-term multi-organ test system for carcinogens has been proposed in which rats are treated by multiple carcinogens to cause initiation in several target tissues followed by administration of the test chemical for 12 weeks. Endpoints that lend themselves to quantitation include preneoplastic lesions in the liver, thyroid, lung, forestomach, urinary bladder, and esophagus.
Over the years it has become apparent that there is no panacea for identifying potential human carcinogens, there is no absolute reference for carcinogenicity, and virtually no human surrogate is ideal or immune to criticism. Equivocal results can occur in any animal model no matter how well a carcinogenicity study is designed. In the last analysis, carcinogenicity data are most relevant to the test model employed. The greatest potential of the various “short-term” organ-specific animal carcinogenesis models is in understanding the underpinnings of the carcinogenic process.
When used with potent carcinogens, a preneoplastic or neoplastic response is often observed in a few weeks, hence the designation “short term.” However, for less potent carcinogens and for non-carcinogens, tests are typically conducted for several months. In the case of the mouse skin-painting model, lifetime studies may sometimes be conducted to assess carcinogenicity. Because of the sharp contrast in the study duration for these so-called “short-term” in vivo test models relative to truly “short-term” (days or weeks) in vivo test systems, they have more recently been referred to as “medium-term” bioassays for carcinogens, indicating a study shorter than the conventional 2-year rodent carcinogenicity study.
“Short-term” or “medium-term” in vivo carcinogenesis animal models consist of an intact animal capable of chemical metabolism (activation and detoxification), complex tissue and hormonal interactions, and possessing repair mechanisms, all of which could influence the ultimate expression of carcinogenic responses to chemical exposure.
Various investigators have proposed specific “short-term” or “medium-term” in vivo rodent models to supplant the more costly chronic rodent carcinogenicity studies (bioassays). In those instances where appropriate validation studies have been conducted, these models have shown unacceptable concordance with results obtained using the “gold standard” chronic rodent carcinogenicity test. Few individuals seem to have considered simple questions such as: Why would a short-duration lung tumor assay system be expected to identify chemicals that produce renal cancer in a chronic rodent study? Despite this unfortunate shortcoming, these alternative in vivo carcinogenicity models can play an important role in defining chemical carcinogenesis.
First, in vivo “short-term” tests are useful in defining the nature of the carcinogenic response observed in a chronic rodent carcinogenicity study. For specific target tissues, they can identify whether a chemical is an initiator or promoter, and they can help define the relative potency of the carcinogen. Second, they can help elucidate the mechanistic basis of carcinogenesis. Third, when used as part of a battery or tier approach to carcinogen testing, they help set priorities for more extensive carcinogenicity testing of chemicals.
3.2. Data Evaluation and Interpretation
Interpretation of results from human epidemiological studies and animal bioassays to identify carcinogenic agents has often proven difficult and controversial. Humans are rarely exposed to only one potential cancer-causing agent in their lifetime, and the amount and duration of that exposure may be difficult or impossible to quantify rigorously. Many years may intervene between exposure to a potential carcinogen and ultimate development of neoplasia, making accurate assessment of cause and effect almost impossible. Relative to man, rodents infrequently develop prostate, colon, pancreatic, cervical, and uterine carcinomas, but develop lung, mammary, hematopoietic, and skin tumors at similar rates (Table 5.13). Neoplasms of the liver, kidney, and thyroid are much more frequent in rodents than in humans.

Despite such limitations, epidemiological studies that clearly show an association between a given chemical exposure or lifestyle habit and an enhanced rate of a specific cancer are regarded as the most relevant method for identification of human carcinogens. While animal bioassays have proven useful for identification of agents that can cause cancer in the laboratory rodent, they only identify an agent as potentially hazardous to human health. Additional facts and factors must be considered in classifying such an agent as a likely human carcinogen (see Statistical Assessment of Toxicologic Pathology Studies, Chapter 30).
The current approach for assessing the scientific relevance of either epidemiological or animal bioassay results to human health risk involves a “weight-of-evidence” procedure in which national and international panels of expert scientists from several disciplines examine all available information on the suspect agent and reach a consensus opinion. Included in this analysis are the strength of the epidemiological evidence, the dose– response curve of the animal response, comparative species metabolism and ability to extrapolate between species, likely mechanism of cancer induction for the agent in question, the genotoxicity of the agent, the amount of the agent in the environment, and the number of people potentially exposed to the agent. Based on this type of analysis, so far more than 100 agents have been classified as known human carcinogens by the International Agency for Research on Cancer (IARC) and the US Health and Human Services Annual Report on Carcinogens (ROC); virtually all are carcinogenic in animals (Table 5.1).
Due to limitations in the animal studies, there remains a lack of certainty in determining if a chemical is not carcinogenic to either humans or animals. The interpretation of animal carcinogenicity results has proven difficult and controversial because there are concerns about adequacy of the rodent bioassay to represent the potential human risk.
The central issues for interpretation are those data dealing with inter-species and low-dose extrapolation. For example, the chemical induction of renal tumors in the male rat by some chemicals appears dependent on a pathway involving the deposition of α2u-globulin in the tubular epithelium;α2u-globulin is not found in humans. Thereby, there is less concern about potential carcinogenic effects of such chemicals in humans when they induce only kidney tumors in male rats by that specific mode of action.
Another example is the occurrence of thyroid neoplasms in rats treated with non-genotoxic enzyme-inducing agents that in turn increase the amount of UDP-glucuronyl transferase, which binds to thyroxin (T4). Because of the excreted and lower circulating levels of T4, thyroid stimulating hormone (TSH) is released and this causes secondary proliferation (hyperplasia and neoplasia of the thyroid).
The high quality of the pathology evaluation for both neoplastic and non-neoplastic lesions is a result of rigorous peer-review (see Peer Review and Pathology Working Groups, Chapter 17). The final interpretation of study results is likewise subjected to intensive peer review, and any carcinogenic response is categorized according to NTP levels of evidence (Table 5.14) of carcinogenic activity. All of the above has resulted in markedly improved data, but has required a substantial expenditure of time and money.

There are positive and negative attributes of the contemporary NTP 2-year rodent carcinogenicity study. On the positive side, the test as performed currently is a thoroughly conducted and peer-reviewed assessment of carcinogenicity and chronic toxicity for the chemical under study. The study results are reasonably interpreted, and provide a better basis for risk assessment than previously designed rodent “bioassays” permitted. All data are published to allow for close scrutiny of the scientific basis for each interpretation.
To date, the 2-year rodent carcinogenicity study appears to be the single best system for identifying carcinogenic potential. Not only has the 2-year rodent carcinogenicity test been sufficiently sensitive in identifying known human carcinogens, it has also identified carcinogens such as aflatoxin, 4-aminobiphenyl, bis(chloromethyl)ether, diethylstilbestrol, melphalan, mustard gas, and vinyl chloride prior to discovery of their carcinogenicity in humans.
Disadvantages of the chronic rodent carcinogenicity test relate primarily to the high cost of each study and to the long time interval required to obtain results. The high costs make the studies too expensive to repeat, thereby obviating the principle of reproducibility demanded by good science. However, in 70 “near-replicate” comparisons, good reproducibility of positivity, target site, and carcinogenic potency was found by comparing published studies done in rats, mice, or hamsters.
The identification of rodent carcinogens on the basis of repeated exposure to high doses of chemical has been criticized as not realistic with respect to human exposure situations in all cases, although clear exceptions exist. Another criticism of the current 2-year rodent carcinogenicity study is that it does not define the mechanisms of any observed carcinogenesis. However, this criticism seems unfounded in that the “standard” chronic rodent carcinogenicity test is not designed to define mechanisms of carcinogenesis. While cost and lack of evidence of reproducibility continue to be valid criticisms of the chronic rodent carcinogenicity test, it still remains the best test system for definitively identifying potential human carcinogens.
An important issue relative to the chronic rodent carcinogenicity test is how the results are interpreted and used. The studies determine if a particular chemical under specific conditions cause cancer in mice or rats. Using judgment and scaling in assessing the significance of the carcinogenicity findings should qualify even this endpoint. For example, a chemical that causes an unequivocal increase in malignant neoplasia in multiple sites and in both sexes of rats and mice should be given different consideration than a chemical that causes a marginal increase of a benign neoplasm whose background incidence is normally quite variable in one sex of one species. This approach is referred to as the “weight-of-evidence” method of interpretation, and it goes further than just evaluating neoplasm incidence data.
Also included in the “weight-of-evidence” method for assessing potential carcinogenic risk to humans is consideration of the nature of the dose–response curve, the pharmacokinetics of the chemical, whether the maximum tolerated dose (MTD) was exceeded in the carcinogenicity study, comparative species metabolism, route of administration, genotoxicity, cytotoxicity, and many other factors (see Pharmacokinetics and Toxicokinetics, Chapter 2).
The judgment of whether a chemical poses a carcinogenic hazard should be made in light of the total evidence provided by all sources of available relevant information. The MTD concept continues to be one of the most debated and controversial issues in toxicology. Studies that are negative for carcinogenicity and were conducted with doses below the MTD leave doubt about the adequacy of the test. One never knows if the animals were sufficiently exposed to the chemical.
Studies conducted at or above the MTD may compromise host homeostasis to such a degree that a positive result has little relevance to human exposure situations. Because it is not always possible to determine what the MTD will be prior to exposure of the animals over much of their lifespan, the real problem comes with how the study results are used in risk assessment. Thus, conclusions regarding carcinogenicity or lack thereof as determined in chronic rodent tests must be qualified as occurring under conditions of the specific study in question. The limitations of the particular study should be pointed out as they relate to MTD, the original design and purpose of the study, study conduct, confounding toxicity, and other study conditions.
In addition, some standardized approach for describing the reasoning used to arrive at a conclusion about the carcinogenicity of a chemical is desirable conceptually and for consistent interpretation. For this purpose, categorization of the observed response using levels of evidence (see Table 5.14) is helpful.
3.3. Genotoxic and Non-Genotoxic Carcinogens
Supportive of the arguments for the mutational hypotheses of cancer causation are the correlation between mutagenicity and carcinogenicity, the correlation between faulty DNA repair mechanisms and some cancers, and the heritable nature of neoplastic transformation. While the correlation between mutagenicity and carcinogenicity is not perfect, many chemical carcinogens are mutagenic either alone or after metabolic activation.
Because it is well known that some mutagens are not carcinogens and some carcinogens are not mutagens, invocation of the association between mutagenicity and carcinogenicity necessarily must be qualified. Practical considerations logically compel us to admit that no single mutagenicity test or even battery of mutagenicity tests would be expected to reflect the complexity and diverse mechanisms of carcinogenesis. Nonetheless, the association between mutagenicity and carcinogenicity, where it occurs, is intuitively appealing as a potential mechanism for the genesis of some cancers.
Endogenous hereditary defects in mutational repair in the face of exposure to environmental mutagenic factors are known to lead to some cancers. Most notable is the occurrence of carcinomas and melanomas of the skin in xeroderma pigmentosum patients who sustain mutations caused by ultraviolet radiation. Because a constellation of biochemical abnormalities may be present in the same cells that have a defective excision repair of pyrimidine dimers, it cannot be adamantly stated that the observed skin cancers result exclusively from the accumulation of mutations resulting from deficient DNA repair. However, the association between defective DNA repair and carcinogenesis in xeroderma pigmentosum patients lends support to the somatic mutation hypothesis of cancer formation.
Non-Alkylating Genotoxic Agents
This class of carcinogen directly substitutes for exocyclic amino groups of nucleosides. The substitution occurs either through oxidative mechanisms or by direct electrophilic attack, and results in a change of base pairing because of deamination or a shift in tautomeric (structural isomer) equilibrium. Examples include nitrous oxide (NO), which causes oxidative deamination, and formaldehyde, which forms crosslinks within DNA. Formaldehyde also causes hydroxymethyl adducts, and thus may also act as an alkylating agent.
Alkylating Genotoxic Agents
Alkylating chemical carcinogens either interact directly with cellular genomic material (direct acting carcinogens) or must first be metabolized by the host to a reactive species (indirect acting carcinogens). Examples of direct alkylating agents include methylnitrosurea, ethylnitrosourea, methylmethane sulfonate, and ethylmethane sulfonate. Indirectly acting alkylating agents require metabolic activation to an electrophilic intermediate and include chemicals such as diethylnitrosamine, benzo[α]pyrene, nitrofurans, nitroquinoline oxide, ethylene dibromide, ethylene dichloride, N-acetyl-2-aminofluorene, and dimethylhydrazine.
Pathways for activation involve cytochrome P450, reduction with NADPH diaphorase, reaction with glutathione, or oxidation mediated by peroxidases or via prostaglandin synthetase. For specifics regarding metabolic activation of different types of chemical carcinogens, the reader is encouraged to consult cited reference material; see also Biochemical and Molecular Basis of Toxicity (Chapter 1). For both direct and indirect acting agents, the reactive form of the carcinogen is an electron-deficient species (electrophilic form) that interacts non-enzymatically with electron-rich or nucleophilic molecular sites in the cell.
These nucleophilic sites are not limited to DNA but also include RNA and cellular protein. Thus, the reaction is not specific for genomic material or for nucleic acids. The interaction of electrophilic forms of the carcinogen with host cellular material results in the formation of covalent adducts (addition products). These interactions take place primarily between the electrophilic form of the carcinogen and the nitrogen, oxygen, and sulfur atoms in the cellular macromolecules.
Adducts formed may be small, as is seen with simple alkylating agents, or large, so-called “bulky adducts,” as occur with polycyclic aromatic hydrocarbons. In either case, configurational or conformational changes occur in the DNA, which can lead to steric hindrance and result in infidelity of DNA replication.
The electrophilic forms of carcinogens react at numerous macromolecular cellular sites and produce adducts and mutations throughout the genome. Some of these mutations may be lethal. Non-lethal reactions with cellular targets are the most relevant for carcinogenesis in that initiation occurs when one of these is in a critical genomic site. Non-lethal alteration in the somatic cell genotype is consistent with the heritable change that is observed in cancer cells.
Evidence of alteration of the cellular genome is supported by the observation of chromosomal abnormalities and altered gene expression in cancer cells. Furthermore, adduct formation at DNA nucleophilic sites such as phosphate residues and hydrogen-bonding sites between base pairs can also result in miscoding or in sufficient distortion of DNA structure to result in infidelity of DNA replication. Because DNA damage (adduct formation) can be repaired efficiently by cellular enzymes, it is necessary for cell proliferation to occur prior to repair of the DNA damage in order for a heritable change to occur. Once a round of cell proliferation takes place, the genomic alteration is said to be “fixed” and the affected cell(s) is initiated. The round or rounds of cell replication that serve to fix the molecular lesions may occur de novo, may be induced by the inherent toxicity of the chemical carcinogen, or may be induced by the promoting activity of the chemical carcinogen.
3.4. Risk Assessment
Considering a risk assessor is required to estimate the dose of an environmental carcinogen that will lead to an increased lifetime probability for cancer of one in a million, continued efforts to accumulate and utilize relevant information are necessary to make scientifically sound estimates. These estimates must also reflect the many endogenous and exogenous factors that influence cancer development, including metabolism of carcinogens, DNA repair capacities, variable genomic stability among animal species, inherited predispositions, immune status, hormones, and our environment.
Cancer risk assessment historically has relied upon classic epidemiology, results from chronic exposures of rodents to potential carcinogens, and mathematical models of these findings. The process of carcinogenesis in rodents is a temporal sequence that represents months in rodents, as opposed to many years in humans. The field of risk assessment has been forced to be cautious mainly because of the limited knowledge of the complex pathogenesis of cancer. Public health strategies are now based upon the premise that reduction or prevention of exposure to cancer causing factors, whether endogenous or exogenous, will decrease the incidence of cancer.
Risk assessment requires numerical estimates of human health risks, and numerous mathematical models have been developed based on animal bioassays, human exposure data, and epidemiology studies to predict a tumor response in a population. These models are based on the theories of multistage carcinogenesis, and take into account initiation, proliferation, multiple genetic alterations, preneoplastic lesions, and the monoclonality of cancer development. The models are only as good as the input data, and often have uncertainties in their predictive value because of factors such as variability of response in outbred populations, sensitivity in measuring tissue dose, and for low-dose extrapolations. These models are useful in determining allowable levels of human exposures, but it should be remembered that they are often based on our imprecise knowledge. As our scientific understanding increases, these “provisional” models will be adjusted to better represent the risks.
Epidemiologic studies that clearly show an association between chemical exposure and an increased risk for the development of cancer provide the strongest evidence for identifying human carcinogens. For most suspected carcinogens, it is often difficult or impossible to quantify the amount and duration of exposure in humans or even to associate the exposure with a disease that may not appear for decades. The animal bioassay identifies agents that are potentially hazardous to human health and must be considered along with as many scientifically-based analyses as are known, such as the dose– response relationship, comparative species metabolism, ability to extrapolate between species, mechanisms of cancer induction, genotoxicity of the agent, amount of agent in the environment, number of people exposed, and strength of the epidemiologic evidence in order to estimate the likelihood that a chemical is carcinogenic in humans (see Pathology Issues in the Design of Toxicology Studies, Chapter 19).
Retrospective or prospective epidemiological methods to study human populations using existing historical records associated with known cases of neoplasia have demonstrated the association of cigarette smoking with lung cancer, and exposure to asbestos with mesotheliomas.
3.5. Molecular Epidemiology
Proto-oncogene and tumor suppressor gene mutation assays have become a popular tool for investigating tumor etiology in humans and rodents mainly because mutations tend to be chemical specific (see The Application of Toxicogenomics to the Interpretation of Toxicologic Pathology, Chapter 11). Because the inactivating mutations of the p53 gene are primarily missense mutations (90%) and there are many “hotspots,” it is “well suited for this form of molecular archaeology” in examining human and animal tumors. Study of the patterns of oncogene activation in spontaneous versus chemically-induced rodent neoplasms has provided data that suggest that the molecular lesions associated with chemically-induced cancer are sometimes different from those documented in spontaneous cancer. Furthermore, the patterns of oncogene activation in several rodent model systems appear to be carcinogen-specific, are consistent with known or expected DNA adduct formation, and in some cases are similar to patterns of oncogene activation documented in human neoplasms.
Mutations in the ras gene from skin tumors in sun-exposed areas appear to be induced by UV irradiation; K-ras mutations in human lung and colon tumors may be the result of adduct formation due to carcinogens from cigarette smoke; and aflatoxin B1-associated human liver tumors appear to have a specific mutation at codon 249 of the p53 gene. There is epidemiological evidence suggesting that for some solvent-associated human leukemias, the exposure was associated with an increased risk of developing a tumor with ras activation.
The analysis of subpopulations of humans at risk with unique genetic alterations in tumors may lead to the identification of more human carcinogens. Moreover, animal studies, such as in the mouse liver tumor models, may help further define these associations and elucidate the effects of dose.
It has been proposed that the carcinogen-specific patterns of ras gene mutation observed in tumors reflect the mechanisms of carcinogenesis, and some patterns of H-ras mutations in mouse liver tumors suggest that they occur from direct chemical–ras gene (genotoxic) interactions.
The B6C3Fl mouse liver is a model in which many genotoxic and non-genotoxic chemicals induce hepatocellular tumors. H-ras activation is a common event in tumorigenesis, and the frequency and pattern of activating mutations are compared between tumors that are chemically-induced and spontaneous to help in determining possible mechanisms by which chemicals induce tumors. Some genotoxic compounds cause mutations that can be explained by the type of DNA adducts formed and some non-genotoxic carcinogens induce tumors that have the same mutational spectra as spontaneous tumors, suggesting that these chemicals act by promoting the spontaneously initiated hepatocytes.
Molecular epidemiology of cancer is the study of molecular alterations, primarily mutations, in investigating the causative agents of cancer, as well as identifying individual cancer risk. The objective to identify cancer-causing agents based on the occurrence of predictable molecular alterations that are found in the neoplasm is intriguing. It is based on the hypothesis that there are carcinogen-specific patterns of mutations that reflect direct interactions of carcinogens with cancer genes. For example, lung and colon cancers from people who smoke tend to have a specific mutation in the ras oncogene or p53 tumor suppressor gene (i.e., mostly a G to T nucleotide base substitution) and that this mutation is likely due to direct interaction of the carcinogen in smoke benzo[α]pyrene with DNA.
Such chemical-specific mutational profiles (or “molecular signatures”) have been used to establish a causal nature between particular genetic events in tumors and carcinogen, such as neoplasms associated with exposure to radon, aflatoxin B1, vinyl chloride, and the nitrosamines. The strongest evidence for linkage between a cancer-causing agent and a specific type of neoplasm is that of the CC to TT double base changes observed in skin neoplasms of man and animals. This mutation is consistent with the predicted UV-induced damage of dipyrimidine dimers. In liver tumors from persons living in geographic areas with a high exposure to aflatoxin B1 there is a frequent mutation at the third nucleotide pair of codon 249 in the p53 gene, suggesting the mutation is chemical-specific and imparts a specific growth or survival advantage to the mutated liver cells.
Animal studies have confirmed that there are certainly chemical-specific mutational profiles in neoplasms; however, there are many examples where the mutational profile varies by strain (Table 5.15), species, dose, or dosing regimen. For example, diethylnitrosamine, a strong, cross-species hepatocarcinogen, will induce liver neoplasms in mice, rats, and rainbow trout, but the frequency and type of ras mutation in the neoplasm varies widely, and the mutations are not simply a reflection of direct DNA interaction.

In some studies, in vitro mutation assays are a poor predictor of liver tumor mutation profiles in the mouse. In this complex process, carcinogens might also be influencing events such as DNA repair, oxidative DNA damage, methylation, cell death, proliferation, and/or a hypermutable state. Other possible mechanisms by which carcinogen treatment could affect ras mutation profile in tumor DNA include those caused by indirect oxidative DNA damage via oxygen or lipid radicals produced during carcinogen metabolism or cytotoxic injury; a selective growth promotion or death of tumor cells with a specific ras genotype; or a hypermutable state induced by the carcinogen (Table 5.11). Other factors that can affect the profile of ras mutations found in mouse liver tumors, and thus add an additional level of complexity in the evaluation of chemical genotoxicity in tumor DNA, include genetic background (strain) of mouse, dose, dosing regimen, and time of sampling. In some cases when humans and rodents are exposed to the same mutagenic carcinogen, such as vinyl chloride, the specific cells or tissue at risk and specific mutations differ.
Using tumor DNA from the National Toxicology Program (NTP) carcinogenesis testing program to detect dominant transforming genes, patterns of oncogene activation have been documented for spontaneous and chemically-induced neoplasms in Fischer 344 rats and in B6C3Fl mice. Results to date show a low frequency (about 3%) of activated oncogenes in spontaneous neoplasms of the Fischer 344 rats.
In contrast, a variety of epithelial neoplasms induced by benzidine congeners as well as lung tumors induced by tetranitromethane inhalation have shown a high frequency of oncogene activation. In the B6C3F1 mouse there is a high frequency (56%) of oncogene activation in spontaneous liver tumors and a lower frequency (8%) in spontaneous non-liver tumors. The oncogenes in spontaneous liver tumors in the B6C3F1 mouse most frequently involve activation of the H-ras proto-oncogene by point mutations in the sixty-first codon.
In chemically-induced hepatic neoplasia in this mouse hybrid, there is also a high frequency of oncogene activation, but the pattern of oncogene activation differs from that observed in spontaneous liver tumors. In addition to a “background” level of mutations in the sixty-first codon identical to those observed in spontaneous hepatocellular neoplasms, additional point mutations were documented in codons 12 or 117 of the H-ras proto-oncogene in addition to the activation of K-ras and N-ras oncogenes in chemically treated mice that had an unequivocal increase in hepatocellular tumors.
Molecular epidemiologic studies aimed at identifying an individual’s risk of developing cancer have found that persons with germline mutations in cancer genes (i.e. BRCA1 or BRCA2) or variations (polymorphisms) of carcinogen-metabolizing enzyme activities (i.e., cytochrome p450s or glutathione-S-transferases) or DNA repair capacities can be at increased risk of developing neoplasia in their lifetime. High-throughput analyses to examine single nucleotide polymorphisms (SNPs) are being used to search for biomarkers of cancer-risk individuals, and some of this information is being used to help people to take preventive measures to decrease their risk of developing cancer.
The most frequently identified activated oncogenes detected in chemically-induced neoplasms belong to the ras gene family. Ras oncogenes were originally detected and isolated by transfection of NIH3T3 cells (immortalized mouse fibroblasts) using DNA from tumors; this method of detection has been replaced by newer molecular techniques. Cloning of these genes from the tumor DNA revealed that many were H-ras, Kras, or N-ras genes that differed from their proto-oncogene homologs by a single point mutation located in a specific codon. Thus, when adolescent rats are given a single exposure to nitrosomethylurea (NMU), mammary tumors result that have an activated H-ras oncogene with a G–A transition (mutation) in the twelfth codon.
G–A transitions are consistent with the Q6- methyl guanine adduct that is formed by methylating agents such as methyl-nitrosourea (MNU), and such mutations would be expected to lead to nucleotide mispairing during DNA replication. The altered DNA would be expected to give rise to an abnormal protein product that could theoretically alter cell growth or differentiation.
MNU is very labile, with an estimated biological half-life of a few minutes. Thus, it is likely that the effect produced by this chemical carcinogen occurred as an early event in the carcinogenic process. Because hormone-stimulated cell division in the mammary tissue is also necessary for the development of mammary neoplasia in this model, it is unlikely that activation of the H-ras gene is sufficient in and of itself for the production of mammary neoplasia. Another example of association of an activated H-ras gene with neoplasia is seen in the carcinomas produced in mouse skin by initiation with 7,12- dimethylbenz[α]anthracene (DMBA) followed by promotion with phorbol ester. In this instance there is an A–T transversion (mutation) in the sixty-first codon of the H-ras gene. Once again, the oncogene activation produced by the DMBA treatment was not sufficient to cause carcinoma development but required promotion by phorbol ester to obtain cancer. Rodent test systems used for the in vivo detection of chemical carcinogens often depend on demonstration of an increased incidence of common neoplasms or on the induction of novel neoplasms in chronically treated animals.
4. CONCLUSIONS
Research is contributing to our better understanding and interpretation of in vivo and in vitro findings of environmentally related carcinogenesis. Studies are identifying the many similarities in the molecular pathogenesis of cancer between rodents and humans. The new technologies will continue to contribute towards our improved understanding of human risks of disease and the betterment of human health.
Although cancer research appears to progress at a snail’s pace, take heart that overall we are making great strides in preventing and treating cancer. The animal models are at least in part responsible for the steady increases in the average American’s lifespan seen over the past three decades.
Acknowledgments
We are grateful to Dr Stephen Mastorides for his contributions to previous editions of the Handbook of Toxicologic Pathology, some of which remain with permission in this updated chapter. We also thank Elizabeth Ney of NIEHS for her assistance generating the tables and figures. This research was supported (in part) by the Division of the National Toxicology Program of the NIH, National Institute of Environmental Health Sciences.
This chapter may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH); however, the statements, opinions, or conclusions contained herein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the United States government.
SUGGESTED READING
Cancer Bioassays
Beyer, L.A., Beck, B.D., Lewandowski, T.A., 2011. Historical perspective on the use of animal bioassays to predict carcinogenicity: evolution in design and recognition of utility. Crit. Rev. Toxicol. 41, 321–338.
Boorman, G.A., Maronpot, R.R., Eustis, S.L., 1994. Rodent carcinogenicity bioassay: past, present, and future. Toxicol. Pathol. 22, 105–111.
Borowsky, A.D., Munn, R.J., Galvez, J.J., Cardiff, R.D., Ward, J.M., Morse 3rd, H.C., Kogan, S.C., Aldape, K.D., Louis, D.N., Bosenberg, M.W., 2004. Mouse models of human cancers (part 3). Comp. Med. 54, 258–270.
Boyland, E., 1969. The correlation of experimental carcinogenesis and cancer in man (review). Prog. Exp. Tumor Res. 11, 222–234.
Cohen, S.M., Arnold, L.L., 2011. Chemical Carcinogenesis. Tox. Sci. 120 (S1), S76–S92.
Farber, E., 1984. Cellular biochemistry of the stepwise development of cancer with chemicals. Cancer Res. 44, 5463–5474.
Hoenerhoff, M.J., Hong, H.H., Ton, T.V., Lahousse, S.A., Sills, R.C., 2009. A review of the molecular mechanisms of chemically induced neoplasia in rat and mouse models in National Toxicology Program bioassays and their relevance to human cancer. Toxicol. Pathol. 37, 835–848.
Huff, J., Haseman, J., 1991. Long-term chemical carcinogenesis experiments for identifying potential human cancer hazards: collective database of the National Cancer Institute and National Toxicology Program (1976–1991). Environ. Health Perspect. 96, 23–31.
Huff, J., Cirvello, J., Haseman, J., Bucher, J., 1991. Chemicals associated with site-specific neoplasia in 1394 long-term carcinogenesis experiments in laboratory rodents. Environ. Health Perspect. 93, 247–270.
Huff, J., Jacobson, M.F., Davis, D.L., 2008. The limits of two-year bioassay exposure regimens for identifying chemical carcinogens. Environ. Health Perspect. 116, 1439–1442.
Maronpot, R.R., Boorman, G.A., 1996. The contribution of the mouse in hazard identification studies. Toxicol. Pathol. 24, 726–731.
Pritchard, J.B., French, J.E., Davis, B.J., Haseman, J.K., 2003. The role of transgenic mouse models in carcinogen identification. Environ. Health Perspect. 111, 444–454.
Tennant, R.W., Margolin, B.H., Shelby, M.D., Zeiger, E., Haseman, J.K., Spalding, J., Caspary, W., Resnick, M., Stasiewwicz, S., Anderson, B., Minor, R., 1987. Prediction of chemical carcinogenicity in rodents from in vitro genetic toxicity assays. Science 236, 933–941.
Molecular Carcinogenesis
Fearon, E.R., Vogelstein, B., 1990. A genetic model for colorectal tumorigenesis. Cell 61, 759–767.
Hanahan, D., Weinberg, R.A., 2011. Hallmarks of cancer: the next generation. Cell 144, 652–674.
Hussain, S.P., Hofseth, L.J., Harris, C.C., 2001. Tumor suppressor genes: at the crossroads of molecular carcinogenesis, molecular epidemiology and human risk assessment. Lung. Cancer 34, S7–S15.
Kinzler, K.W., Vogelstein, B., 1996. Lessons from hereditary colorectal cancer. Cell 87, 159–170.
Loeb, L.A., 1994. Microsatellite instability: marker of a mutator phenotype in cancer. Cancer Res. 54, 5059–5063.
Malarkey, D.E., Parker, J.S., Turman, C.A., Scott, A.M., Paules, R.S., Maronpot, R.R., 2005. Microarray data analysis of mouse neoplasia. Toxicol. Pathol. 33, 127–135.
Vogelstein, B., Kinzler, K.W., 2004. Cancer genes and the pathways they control. Nat. Med. 10, 789–799.
Wood, L.D., Parsons, D.W., Jones, S., Lin, J., Sjo¨blom, T., Leary, R.J., Shen, D., Boca, S.M., Barber, T., Ptak, J., Silliman, N., Szabo, S., Dezso, Z., Ustyanksky, V., Nikolskaya, T., Nikolsky, Y., Karchin, R., Wilson, P.A., Kaminker, J.S., Zhang, Z., Croshaw, R., Willis, J., Dawson, D., Shipitsin, M., Willson, J.K., Sukumar, S., Polyak, K., Park, B.H., Pethiyagoda, C.L., Pant, P.V., Ballinger, D.G., Sparks, A.B., Hartigan, J., Smith, D.R., Suh, E., Papadopoulos, N., Buckhaults, P., Markowitz, S.D., Parmigiani, G., Kinzler, K.W., Velculescu, V.E., Vogelstein, B., 2007. The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113.
Hepatocarcinogenesis
Allen, D.G., Pearse, G., Haseman, J.K., Maronpot, R.R., 2004. Prediction of rodent carcinogenesis: an evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol. Pathol. 32, 393–401.
Christensen, J., Romach, E.H., Healy, L.N., Gonzales, A.J., Anderson, S.P., Malarkey, D.E., Cattley, R.C., Goldsworthy, T.L., 1999. Altered Bcl-2 family expression during nongenotoxic hepatocarcinogenesis in mice. Carcinogenesis 20, 1583–1590.
Hailey, J.R., Haseman, J.K., Bucher, J.R., Radovsky, A.E., Malarkey, D.E., Miller, R.T., Nyska, A., Maronpot, R.R., 1998. Impact of Helicobacter hepaticus infection in B6C3F1 mice from twelve National Toxicology Program two-year carcinogenesis studies. Toxicol. Pathol. 26, 602–611.
Hall, A.P., Elcombe, C.R., Foster, J.R., Harada, T., Kaufmann, W., Knipple, A., Kuttler, K., Malarkey, D.E., Maronpot, R.R., Nishikawa, A., Nolte, T., Schulte, A., Strauss, V., York, M.J., 2012. Liver Hypertrophy: A Review of Adaptive (Adverse and Non-adverse) Changes – Conclusions from the 3rd International ESTP Expert Workshop. Toxicol. Pathol. 2012 Jun 21 [Epub].
Hoenerhoff, M.J., Pandiri, A.R., Lahousse, S.A., Hong, H.H., Ton, T.V., Masinde, T., Auerbach, S.S., Gerrish, K., Bushel, P.R., Shockley, K.R., Peddada, S.D., Sills, R.C., 2011. Global gene profiling of spontaneous hepatocellular carcinoma in B6C3F1 mice: similarities in the molecular landscape with human liver cancer. Toxicol. Pathol. 39, 678–699.
Malarkey, D.E., Devereux, T.R., Dinse, G.E., Mann, P.C., Maronpot, R.R., 1995. Hepatocarcinogenicity of chlordane in B6C3F1 and B6D2F1 male mice: evidence for regression in B6C3F1 mice and carcinogenesis independent of ras proto-oncogene activation. Carcinogenesis 16, 2617–2625.
Maronpot, R.R., Fox, T., Malarkey, D.E., Goldsworthy, T., 1995. Mutations in the ras proto-oncogene: Clues to etiology and molecular pathogenesis of mouse liver tumors. Toxicology 101, 125–156.
Maronpot, R.R., Yoshizawa, K., Nyska, A., Harada, T., Flake, G., Mueller, G., Singh, B., Ward, J.M., 2010. Hepatic Enzyme Induction: Histopathology. Toxicol. Pathol. 38, 776.
Stout, M.D., Kissling, G.E., Sua´rez, F.A., Malarkey, D.E., Herbert, R.A., Bucher, J.R., 2008. Influence of Helicobacter hepaticus infection on the chronic toxicity and carcinogenicity of triethanolamine in B6C3F1 Mice. Toxicol. Pathol. 36, 783–794.
Spectrum of Proliferative Lesions
Bannasch, P., 1986. Preneoplastic lesions as end points in carcinogenicity testing. I. Hepatic preneoplasia. Carcinogenesis 7, 689–695.
Bannasch, P., 1986. Preneoplastic lesions as end points in carcinogenicity testing. II. Preneoplasia in various nonhepatic tissues. Carcinogenesis 7, 849–852.
Cardiff, R.D., Anver, M.R., Boivin, G.P., Bosenberg, M.W., Maronpot, R.R., Molinolo, A.A., Nikitin, A.Y., Rehg, J.E., Thomas, G.V., Russell, R.G., Ward, J.M., 2006. Precancer in mice: animal models used to understand, prevent, and treat human precancers. Toxicol. Pathol. 34, 699–707.
Medina, D., 1988. The preneoplastic state in mouse mammary tumorigenesis. Carcinogenesis 9, 1113–1119.
Nowell, P.C., 1986. Mechanisms of tumor progression. Cancer Res. 46, 2203–2207.
Initiation, Promotion, and Progression
Argyris, T.S., 1985. Promotion of epidermal carcinogenesis by repeated damage to mouse skin. Am. J. Ind. Med. 8, 329–337.
Borzsonyi, M., Day, N.E., Lapis, K., Yamasaki, H. (Eds.), 1984. “Models, Mechanisms, and Etiology of Tumour Promotion.” IARC. France, Lyon.
Hennings, H., Yuspa, S.H., 1985. Two–stage tumor promotion in mouse skin: An alternative interpretation. J. Natl. Cancer. Inst. 74, 735–740.
Clonal Origins
Fialkow, P.J., 1979. Clonal origin of human tumors. Annu. Rev. Med. 30, 135–143.
Nowell, P., 1978. Tumors as clonal proliferations. Arch. Cell Pathol. 29, 145–150.
Cancer Stem Cell Theory (Extended Bibliography)
Aguirre-Ghiso, J.A., 2007. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 7, 834–846.
Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J., Clarke, M.F., 2003. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988.
Al-Hajj, M., Becker, M.W., Wicha, M., Weissman, I., Clarke, M.F., 2004. Therapeutic implications of cancer stem cells. Curr. Opin. Genet. Dev. 14, 43–47.
Bonnet, D., Dick, J.E., 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737.
Conheim, J., 1867. Ueber entzundung und eiterung. Path. Anat. Physiol. Klin. Med. 40, 1–79.
Conheim, J., 1875. Congenitales, Quergestreiftes Muskelsarkon der Nireren. Virchows. Arch. 65, 64.
Donnenberg, V.S., Donnenberg, A.D., 2005. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J. Clin. Pharmacol. 45, 872–877.
Dontu, G., Wicha, M.S., 2005. Survival of mammary stem cells in suspension culture: implications for stem cell biology and neoplasia. J. Mammary Gland. Biol. Neoplasia. 10, 75–86.
Dontu, G., Al-Hajj, M., Abdallah, W.M., Clarke, M.F., Wicha, M.S., 2003. Stem cells in normal breast development and breast cancer. Cell Prolif. 36 (Suppl. 1), 59–72.
Durante, F., 1874. Nesso fisio-pathologico tra la struttura dei nei materni e las genesi di alcuni tumori maligni. Arch. Memori. ed. Osservazioni. di. Chirugia. Practica. 11, 217–226.
Eramo, A., Lotti, F., Sette, G., Pilozzi, E., Biffoni, M., Di Virgilio, A., Conticello, C., Ruco, L., Peschle, C., De Maria, R., 2008. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Diff. 15, 504–514.
Fang, D., Nguyen, T.K., Leishear, K., Finko, R., Kulp, A.N., Hotz, S., Van Belle, P.A., Xu, X., Elder, D.E., Herlyn, M., 2005. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 65, 9328–9337.
Farrar, J.D., Katz, K.H., Windsor, J., Thrush, G., Scheuermann, R.H., Uhr, J.W., Street, N.E., 1999. Cancer dormancy. VII. A regulatory role for CD8þ T cells and IFNgamma in establishing and maintaining the tumordormant state. J. Immunol. 162, 2842–2849.
Kim, C.F., Jackson, E.L., Woolfenden, A.E., Lawrence, S., Babar, I., Vogel, S., Crowley, D., Bronson, R.T., Jacks, T., 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835.
Klein, W.M., Wu, B.P., Zhao, S., Wu, H., Klein-Szanto, A.J., Tahan, S.R., 2007. Increased expression of stem cell markers in malignant melanoma. Mod. Pathol. 20, 102–107.
Kordon, E.C., Smith, G.H., 1998. An entire functional mammary gland may comprise the progeny from a single cell. Development 125, 1921–1930.
Lang, S.H., Frame, F.M., Collins, A.T., 2009. Prostate cancer stem cells. J. Pathol. 217, 299–306.
Lessard, J., Sauvageau, G., 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells Nature 423, 255–260.
Li, C., Heidt, D.G., Dalerba, P., Burant, C.F., Zhang, L., Adsay, V., Wicha, M., Clarke, M.F., Simeone, D.M., 2007. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037.
Liu, S., Dontu, G., Wicha, M.S., 2005. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast. Cancer Res. 7, 86–95.
Lowenberg, B., Terpstra, W., 1998. Maturation hierarchy of leukemic stem cells. Stem cells 16 (Suppl. 1), 85–89.
Monzani, E., Facchetti, F., Galmozzi, E., Corsini, E., Benetti, A., Cavazzin, C., Gritti, A., Piccinini, A., Porro, D., Santinami, M., Invernici, G., Parati, E., Alessandri, G., La Porta, C.A., 2007. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumorigenic potential. Eur. J. Cancer 43, 935–946.
O’Brien, C.A., Pollett, A., Gallinger, S., Dick, J.E., 2007. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110.
Pardal, R., Molofsky, A.V., He, S., Morrison, S.J., 2005. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb. Symp. Quant. Biol. 70, 177–185.
Park, I.K., Qian, D., Kiel, M., Becker, M.W., Pihalja, M., Weissman, I.L., Morrison, S.J., Clarke, M.F., 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305.
Pathak, S., 2002. Organ- and tissue-specific stem cells and carcinogenesis. Anticancer Res. 22, 1353–1356.
Pierce, G.B., 1967. Teratocarcinoma: model for a developmental concept of cancer. Curr. Top. Dev. Biol. 2, 223–246.
Reya, T., Morrison, S.J., Clarke, M.F., Weissman, I.L., 2001. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111.
Ricci-Vitiani, L., Lombardi, D.G., Pilozzi, E., Biffoni, M., Todaro, M., Peschle, C., De Maria, R., 2007. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115.
Schatton, T., Murphy, G.F., Frank, N.Y., Yamaura, K., WaagaGasser, A.M., Gasser, M., Zhan, Q., Jordan, S., Duncan, L.M., Weishaupt, C., Fuhlbrigge, R.C., Kupper, T.S., Sayegh, M.H., Frank, M.H., 2008. Identification of cells initiating human melanomas. Nature 451, 345–349.
Sell, S., 2004. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 51, 1–28.
Sell, S., Pierce, G.B., 1994. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Invest. 70, 6–22.
Shackleton, M., Vaillant, F., Simpson, K.J., Stingl, J., Smyth, G.K., Asselin-Labat, M.L., Wu, L., Lindeman, G.J., Visvader, J.E., 2006. Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88.
Singh, S.K., Hawkins, C., Clarke, I.D., Squire, J.A., Bayani, J., Hide, T., Henkelman, R.M., Cusimano, M.D., Dirks, P.B., 2004. Identification of human brain tumour initiating cells. Nature 432, 396–401.
Soltysova, A., Altanerova, V., Altaner, C., 2005. Cancer stem cells. Neoplasma. 52, 435–440.
Spillane, J.B., Henderson, M.A., 2007. Cancer stem cells: a review. ANZ. J. Surg. 77, 464–468.
Taipale, J., Beachy, P.A., 2001. The Hedgehog and Wnt signalling pathways in cancer. Nature 411, 349–354.
Till, J.E., Mc, C.E., 1961. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 14, 213–222.
Tu, S.M., Lin, S.H., Logothetis, C.J., 2002. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet. Oncol. 3, 508–513.
Welm, B., Behbod, F., Goodell, M.A., Rosen, J.M., 2003. Isolation and characterization of functional mammary gland stem cells. Cell Prolif. 36 (Suppl. 1), 17–32.
Yoo, M., Hatfield, D.L., 2008. The cancer stem cell theory: Is it correct? Mol. Cells 26, 514–516.
Zhang, S., Balch, C., Chan, M.W., Lai, H.C., Matei, D., Schilder, J.M., Yan, P.S., Huang, T.H., Nephew, K.P., 2008. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 68, 4311–4320.