Cancer, or neoplasia, which occurs in one of every four individuals and results in the death of one of every five individuals in the United States, is a complex disease with multiple causes. Many intrinsic and extrinsic factors influence the development of cancer. Intrinsic or host factors include age, sex, genetic constitution, immune system function, metabolism, hormone levels, and nutritional status. Extrinsic factors include substances eaten, drunk, or smoked; workplace and environmental (air, water, and soil) exposures; natural and medical radiation exposure; sexual behavior; and elements of lifestyle such as social and cultural environment, personal behavior, and habits. Intrinsic and extrinsic factors can interact with one another to influence the development of cancer. Because of the physical and emotional suffering associated with cancer and the immense cost to the nation in lost production and income and medical and research expenditures, considerable effort continues to be exerted to understand this complex disease so that strategies can be developed to decrease or prevent its occurrence. Current regulatory guidelines have been crafted to reduce the probability of developing cancer by lowering human exposure to agents identified as potentially capable of causing cancer.
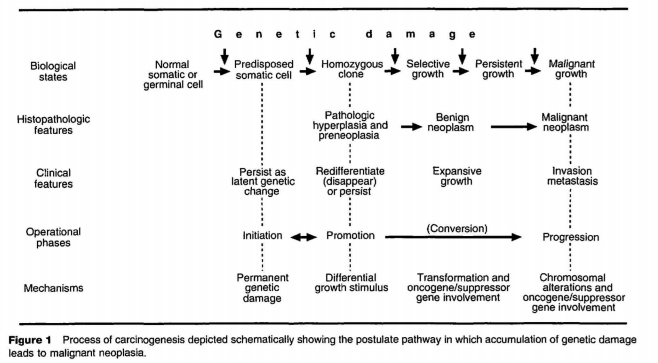
During the past 40 years of cancer research, much information has been generated indicating that cancer is a multistep, progressive disease. Support for this contention is derived from research on epidemiology and population genetics, morphological and clinical study of neoplasms, as well as experimental investigations in animals. Structural studies of biopsy and autopsy tissue samples from humans and animals, particularly experimental animal models of carcinogenesis, have provided important information about this multistep process at the phenotypic level. More recently, molecular biological analyses have confirmed the principles that neoplasms arise from the clonal expansion of a single cell and that during its evolution into a neoplastic mass, it accumulates nonlethal genetic damage, particularly in genes that regulate growth and DNA repair processes. The process of carcinogenesis may take months in experimental laboratory animals and years in humans. Identification of this process early in its evolution enhances the likelihood that intervention strategies such as surgical removal of a benign neoplasm may result in termination of the disease and clinical cure. By the time a neoplasm has progressed to the malignant stage and spread throughout the body, even heroic radiation and chemotherapy combined with surgery are unlikely to result in clinical cure. The process of carcinogenesis may be depicted schematically as in Figure 1 with the various steps along the pathway from normalcy to malignancy characterized by morphological and/or clinical features. It is here that the disciplines of clinical oncology, molecular biology and pathology are utilized to define the location of the specific neoplasm in this progressive cascade.
Nomenclature of Cancer (Neoplasia)
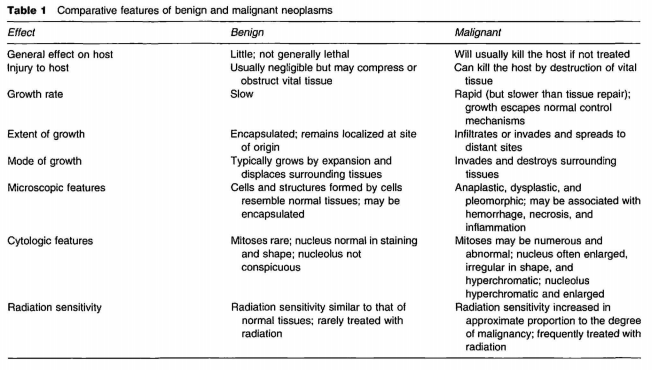
The nomenclature associated with the study of cancer is frequently confusing because a given term often has a relatively narrow as well as a considerably broader definition based on common usage. Carcinogenesis, for example, is narrowly defined as the production of carcinoma but is more commonly used in the broadest possible sense to indicate generation of neoplasms which are new and typically abnormal growths, generally uncontrolled and becoming progressively more serious with time. Neoplasia, meaning ‘new growth’ and often used synonymously with carcinogenesis, refers to the process of development of neoplasms. Two important terms which relate to the clinical behavior and growth characteristics of neoplasms are (1) benign and (2) malignant, characteristic features of which are listed in Table 1. Basically, benign neoplasms are slow-growing, localized growths frequently amenable to surgical removal with a low probability of recurrence. Malignant neoplasms have a more aggressive growth, are locally invasive, sometimes metastasize (spread to distant sites), and are difficult to remove surgically.
Two terms that have both a narrow and a broad definition are (1) tumor and (2) cancer. Tumor broadly refers to any tissue enlargement or swelling, however it is often used synonymously with the term neoplasm. A cancer generally refers to a malignant neoplasm. Unfortunately, the layperson and the professional frequently use tumor and cancer interchangeably alike without qualifying whether it is a benign or malignant process. In other words, if it is said that an individual has a tumor, that individual may have a benign neoplasm (most often the case) but could have a malignant neoplasm if the term ‘tumor’ is being used loosely. If an individual is said to have a cancer, that usually means the individual has a malignant neoplasm but, here again, loose use of the term ‘cancer’ might include any neoplasm, including a benign one. Scientists contribute to the confusion by sometimes indicating that an agent may cause cancer, meaning either benign or malignant neoplasia. Alternatively, they may indicate that an agent is tumorigenic, which could mean that it causes tumors but frequently means that it may also cause malignant neoplasms (cancers). Common and uncritical usage of these terms is so ingrained that attempts to standardize nomenclature have been largely unsuccessful. The least ambiguous terms are ‘benign neoplasm’ and ‘malignant neoplasm’.
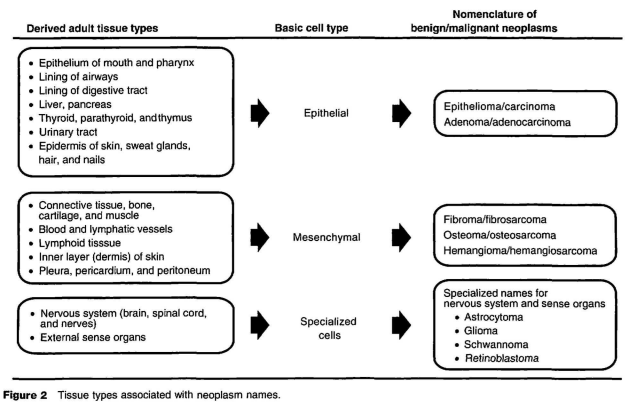
Most neoplasms are classified and named based on (1) the cell or tissue of origin and (2) benign or malignant growth characteristics. There are two basic cell types from which neoplasms may originate: mesenchymal cells and epithelial cells (Figure 2). Mesenchymal pertains to mesenchyma (embryonic connective tissue in the mesoderm) from which adult tissues such as connective tissue, blood and lymphatic vessels, and muscles and bones are formed. Epithelial cells line the internal and external surfaces of the body and form many of the major organs such as liver and lungs. Most epithelial tissues are derived from the embryonic germ layers referred to as entoderm and ectoderm.
There are general guidelines used in naming neoplasms. A benign epithelial neoplasm originating within a glandular tissue is called an ‘adenoma’, having the prefix ‘adeno’ to designate that the origin is one of many glandular tissues and the suffix ‘oma’ to indicate a swelling or tissue enlargement. One or more qualifiers may be added to the name to indicate the tissue of origin and various morphological features as in hepatocellular (liver cell) adenoma, thyroid follicular (forming follicles) adenoma, or renal (kidney) tubular cell adenoma. An adenoma with morphological features resembling finger-like or warty projections would be called a papillary adenoma; with cystic spaces, a cystadenoma; with both of these features, a papillary cystadenoma. Benign mesenchymal neoplasms also utilize the ‘oma’ suffix in their name, as in meningioma, hemangioma, and fibroma. The prefix for mesenchymal neoplasms usually identifies the specific tissue of origin such as meninges (meningioma), blood vessels (hemangioma), or fibrous connective tissue (fibroma). Nomenclature for several benign neoplasms is presented in Table 2.
Malignant epithelial neoplasms are typically called ‘carcinomas’ and qualified by histogenetic origin. Thus, malignant skin neoplasms are called epidermal carcinomas if they arise in the superficial layers or epidermis of the skin. If they are composed predominantly of squamous cells, they are called squamous cell carcinomas; if chiefly basal cells, basal cell carcinomas. Malignant mesenchymal neoplasms are called ‘sarcomas’. Examples of the latter include fibrosarcoma, a malignant neoplasm of the connective tissue; osteosarcoma, a malignant bone neoplasm; and leiomyosarcoma, a malignant neoplasm of the smooth muscle tissue. The nomenclature for several malignant neoplasms is presented in Table 2.
Much of the general confusion surrounding the nomenclature of neoplasms results from numerous exceptions and permutations in the general histogenetic and clinical guidelines for naming neoplasms. Many of these exceptions are deeply ingrained in traditional pathology practice, and attempts at standardization have been largely unsuccessful. Examples are thymoma, lymphoma, melanoma, and neuroblastoma – neoplasms which are generally regarded as malignant despite their benignsounding names and should more properly be called malignant thymoma or thymic sarcoma, malignant lymphoma or lymphosarcoma, malignant melanoma or melanosarcoma, and malignant neuroblastoma, respectively. Other neoplasms are named for their physical attributes such as pheochromocytoma (darkcolored neoplasms typically arising in the adrenal medulla). In addition, some neoplasms were originally named for the person first describing the lesion, and examples such as Hodgkin’s disease of lymphoid tissue and Wilms’ kidney tumor have persisted to this day. Neoplasms composed of mixtures of cells are named accordingly; examples include fibroadenoma, adenosquamous carcinoma, and carcinosarcoma. To complicate matters further there are several tissue alterations that are not neoplasms but have names suggesting that they are: hamartomas (a disorganized aggregate of normal tissue components thought to represent faulty differentiation during embryonic development) and choristomas (focal collections of normal tissue found at an abnormal site such as islands of pancreatic cells in the wall of the stomach). There are also instances in which a neoplasm is histologically considered malignant but clinically benign, such as in basal cell carcinoma of the skin. In addition, localized overgrowths of normal tissue components such as skin tags and vocal cord polyps arc clinically recognized as tumors but are not truly neoplastic.
For brief definitions of various terms associated with carcinogenesis, refer to the glossary at the end of this entry.
Tissue Changes Associated with Carcinogenesis
Quantitative – Hyperplasia and Preneoplasia
Proliferative lesions, which may be classified morphologically as hyperplasia, preneoplasia, benign neoplasia, or malignant neoplasia, represent a continuum of change with considerable overlap rather than discrete morphologic entities (Figure 3). The definitive classification of a given lesion as preneoplasia, benign neoplasia, or malignant neoplasia represents a judgment based on the experience of the diagnostic pathologist and familiarity with the species and tissue in question. These lesions are recognized by their microscopic appearance and effect on surrounding tissues and typically are a localized proliferation or hyperplasia of a specific cell type. Most neoplasms are believed to be derived from the clonal proliferation of a single initiated cell. Usually at somepoint early in the clonal expansion, the differentially proliferating cells become phenotypically distinguishable from the surrounding normal tissue. Although such lesions may not yet have sufficient characteristics to qualify as neoplasms, their recognition early in the process of carcinogenesis has led many to regard them as ‘preneoplastic’.
There is considerable confusion regarding the significance of hyperplasia in the neoplastic process. Hyperplasia is an increase in the number of cells per unit of tissue, typically limited in amount and terminating when the stimulus that evoked it is removed. Different cell types have varying capacities to undergo hyperplasia in response to physiological or pathological stimuli. One of the most difficult judgments, even for the experienced pathologist, is whether an observed hyperplasia is part of the process of cancer development or merely an adaptive or physiologic response not likely to progress to neoplasia. The tissue affected, whether the hyperplasia is diffuse or nodular, the age of the affected individual, the proximate cause of the hyperplastic response, and the growth pattern of the hyperplastic tissues, influences this judgment.
Preneoplasia is a form of hyperplasia (an absolute increase in the number of cells in a tissue). Although not all neoplasms exhibit a preneoplastic change recognizable by the pathologist, in those instances in which presumptive alterations are observed, their occurrence documents that there is a response to tissue insult. Examples of presumptive preneoplastic lesions are presented in Table 3. In those experimental models of carcinogenesis in which preneoplasia is observed, it precedes the occurrence of benign neoplasia. An important feature of preneoplastic lesions is their propensity for reversibility. In some instances a preneoplastic lesion represents the clonal expansion of a cell that has sustained genetic damage so that benign neoplasms arise within the preneoplastic lesion, presumably when one of the preneoplastic cells sustains additional genetic damage, giving it a growth advantage. In other situations, the antecedent change is a localized polyclonal cellular proliferation historically associated with subsequent development of a neoplasm in the same tissue.
A classical example is alcoholic cirrhosis, which in the case of chronic alcohol abuse, leads to multiple, polyclonal areas of liver cell hyperplasia and an increased risk for development of hepatocellular neoplasia. In both preneoplasia and certain forms of hyperplasia, the antecedent lesions typically have a higher rate of cell proliferation than the surrounding norma! cells and, thus, these cells are at increased risk to sustain additional genetic damage and progress to the next stage in the carcinogenic process.
A benign neoplasm is generally a localized expansive growth that compresses adjacent normal tissue but is usually not immediately life threatening unless it physically interferes with normal function, for example, by blocking the intestinal tract or compressing vital areas in the brain. Controversy regarding the significance of benign neoplasia with respect to the development of malignancy is similar to that associated with preneoplastic lesions. A benign neoplasm, the clonal expansion of cells that have sustained some degree of genetic damage, is further along the spectrum of changes that precede the development of malignant neoplasia. In experimental carcinogenesis animal models, malignant neoplasia is not infrequently observed arising from or within a benign neoplasm. Features of benign neoplasms are listed in Table 1.
Malignant neoplasms are rapidly growing, locally invasive tissue proliferations that destroy surrounding tissues and are thus life threatening. They also have the malicious feature of spreading to distant sites in the body via the blood and lymphatic system. Although malignancy develops with greater frequency in association with (1) pathologic hyperplasia and preneoplasia, (2) qualitative alterations in cells, and (3) benign neoplasia than in association with normal tissues, these changes are not necessary precursors to malignancy. In situ carcinomas are malignant neoplasms that originate without evidence of antecedent benign tissue alteration. When precursor lesions are present prior to or concomitant with malignant neoplasia, it is probable that the malignancy is a consequence of the same or similar factors that produced the precursor lesions. Characteristics of malignant neoplasms are listed in Table 1.
Qualitative – Metaplasia, Dysplasia, and Anaplasia
In addition to quantitative increases in certain cells, several qualitative cytological features help allow the morphologic classification of the spectrum of proliferative lesions that may be observed in the process of carcinogenesis. Three frequently used qualitative cytological features are metaplasia, dysplasia, and anaplasia.
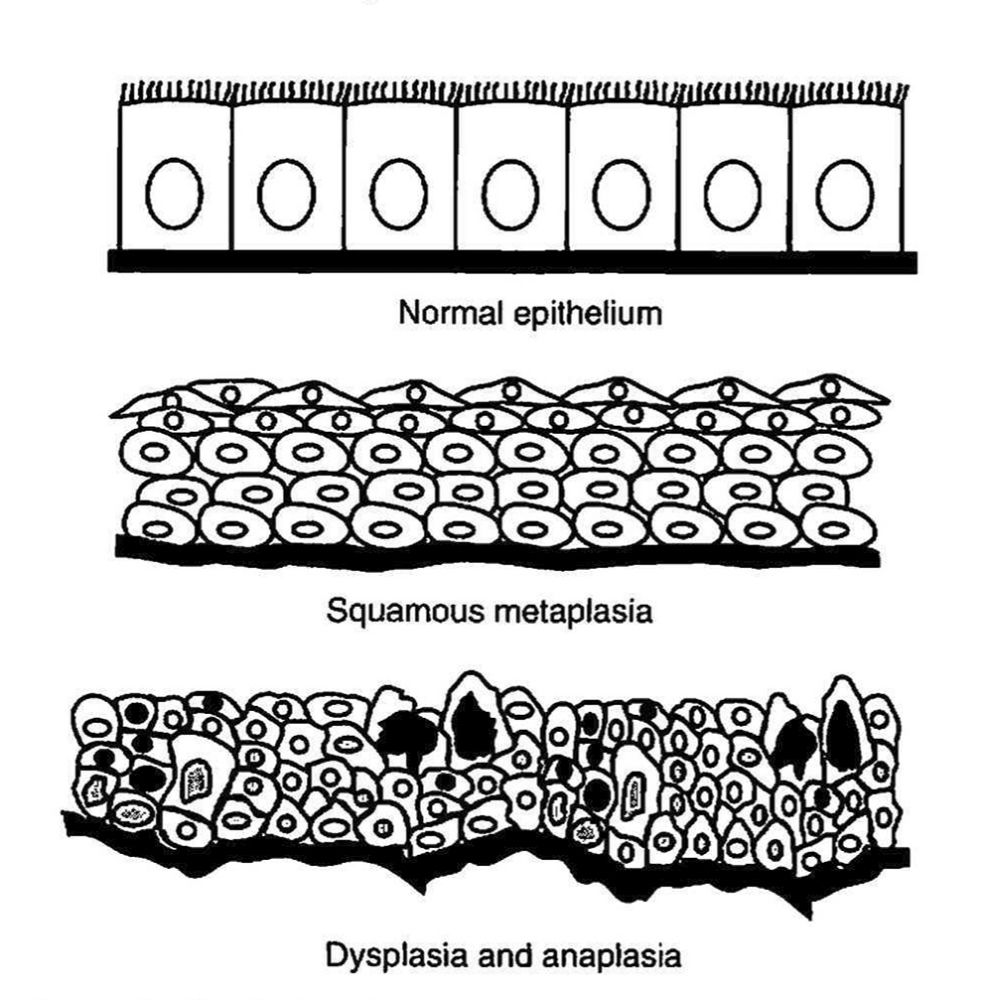
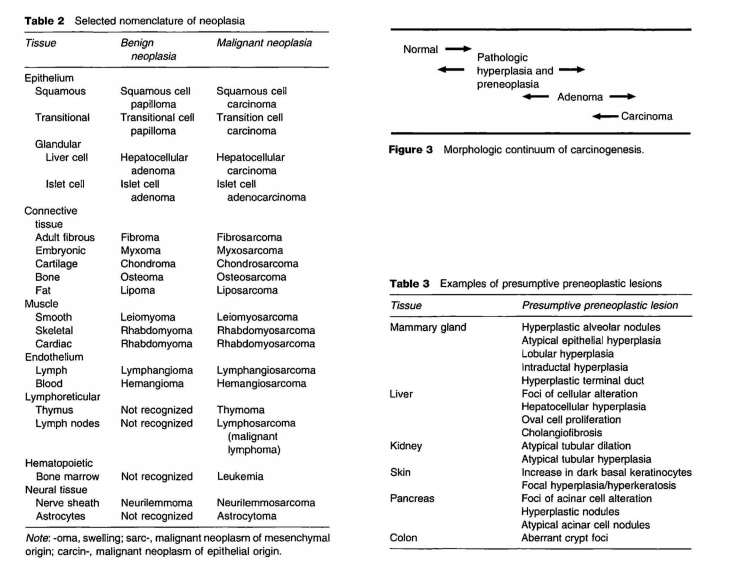
Metaplasia is the reversible substitution of one type of fully differentiated cell for another within a given tissue. A classic example is the replacement of the normal ciliated columnar epithelial cells in the respiratory tract airways by squamous epithelium (Figure 4) in situations in which there is chronic irritation from certain components of inhaled tobacco smoke. While the squamous epithelium is believed to provide functional protection against the irritant properties of the smoke, the loss of the ciliated columnar epithelium results in reduction of the functional capacity of the lungs to clear particulates from the respiratory tract. When the irritation is removed, the squamous epithelium is replaced by normal ciliated columnar epithelium.
Dysplasia is defined as abnormal growth of a tissue with respect to shape, size, and the organization of component cells. Normal cell-to-cell orientations arc disorganized or disrupted, and the cells themselves vary in size and shape (Figure 4). When present, dysplasia may be associated with chronic irritation, occur with metaplasia, and be seen in neoplastic transformation. It is a change that is a hallmark of increased risk for development of neoplasia. Like metaplasia, dysplasia is a potentially reversible tissue alteration. It is also considered in some circumstances as a preneoplastic change.
Anaplasia is a qualitative alteration of cellular differentiation. Anaplastic cells are typically undifferentiated and may bear little, if any, resemblance to mature cells. This feature is considered a hallmark of malignancy.
Staging and Grading of Cancers
In human oncology the experience from collective years of observation of the outcome of many cancers has strengthened the predicitivity of histological grades and clinical staging in prognostication. The purpose of grading and staging a neoplasm is to predict its biological behavior and to help establish an appropriate therapeutic regimen. Grading is a subjective evaluation of morphologic characteristics based on the extent of cellular anaplasia and the degree of proliferation evident from microscopic evaluation. Generally, neoplasms with a high degree of anaplasia, associated specific morphologic patterns of growth, and evidence of numerous mitoses, some of which may be abnormal, are given a high grade of malignancy. Most grading schemes categorize neoplasms into one of three or four grades of increasing malignancy.
Staging of a cancer, which is independent of grading, is an index of the extent to which a cancer has spread in the body. It also provides information regarding the patient’s clinical prognosis, and usually influences the choice of appropriate therapy more than grading. Criteria used for staging neoplasms include the size of the primary neoplasm, the degree to which there is invasion of surrounding normal tissues, whether the cancer has spread to local lymph nodes, and the presence of spread to distant sites in the body. Thus, it is apparent that staging will have a large influence on the therapeutic approach. A small and localized breast cancer would most likely be treated by surgical excision and possibly radiation therapy, whereas a large, infiltrative breast cancer would more likely be treated by mastectomy. If the cancer has spread to lymph nodes or distant sites, more aggressive therapy is implemented.
The ultimate fate of cells or proliferative tissue masses is influenced by the amount of sustained genetic damage. Cells with minimal DNA damage may persist in a latent form, indistinguishable from surrounding normal cells. If such a latent cell sustains additional damage even long after the initial insult, it may then progress further along the pathway to malignancy (see Figure 1). As additional genetic damage occurs, the altered cell population expands and eventually leads to irreversible uncontrolled growth that may or may not be corrected by aggressive medical intervention.
Molecular Basis of Cancer
Multistep Genetic Model of Carcinogenesis
Genetically, the multistage process involves the activation of growth-enhancing protooncogenes, inactivation of the recessive growth-inhibitory tumor suppressor genes as well as epigenetic events that alter gene expression and processes such as those involved in cell death, DNA repair, and methylation (Table 4). Cancer cells frequently contain mutations in multiple genes as well as large chromosomal abnormalities. Since their discovery ~2 5 years ago, more than 100 protooncogenes and ~ 15 tumor suppressor genes have been identified. Protooncogenes were first discovered in cancer-causing animal viruses that carried them. Intense study of these viruses, particularly by Varmus and Bishop in the 1970s, resulted in the discovery that endogenous animal genes had been picked up by virus ancestors and incorporated into the viral genome. Soon thereafter a number of these protooncogenes were identified in both the animal and human genome and later found to play a role in cancer development.
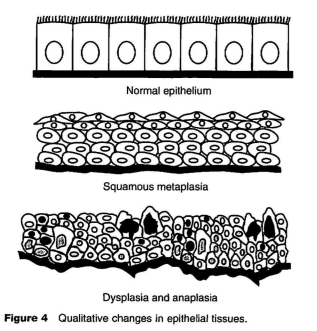
A widely accepted multistep model of carcinogenesis proposed by Fearon and Vogelstein in 1990 serves as the framework for studies in carcinogenesis (Figure 5). By studying multiple benign and malignant colonic neoplasms from individuals with multiple tumors it was found that benign neoplasms harbored mutations in genes such as APC, ras, and p53, and that there were frequently multiple mutations per neoplasm, particularly of the malignant neoplasms. The model describes a progressive acquisition of mutations and it is believed the total accumulation of mutations (at least five to seven) rather than the order is important in the carcinogenic process. New evidence has been published to further refine this model. Recently it has been proposed that some neoplasms are dependent on the continued activation or overexpression of a particular oncogene for maintaining malignant behavior. Others have found that some neoplasms are ‘hypersensitive’ to the inhibitory effects of specific tumor suppressor genes. These findings suggest that the multistage process of carcinogenesis is not simply a summation of individual effects of cancer genes but that some individual cancer genes can override the others (referred to by some as the ‘Achilles heel of cancer’) and they offer new strategies for the prevention and therapy of cancer.

Oncogenes
Among the estimated 25,000 genes in the mammalian genome, there are ~ 100 genes that are classified as oncogenes because activation of these genes appears to be an essential event for the development of many, if not all, cancers. In fact, oncogenes were first discovered by studying genetic alterations in cancers. The term oncogene activation indicates a quantitative or qualitative alteration in the expression or function of the oncogene. The term oncogene is unfortunate since the unaltered (nonactivated) oncogene (usually referred to as a protooncogene) actually serves an essential function in the mammalian genome. That protooncogenes are highly conserved in evolution is evidenced by structurally and functionally similar genes in yeast, earthworms, animals, and humans. The highly conserved nature of protooncogenes is believed to be related to their essential function in normal tissue growth and differentiation. Since their normal function is to control how a tissue grows and develops, it is apparent that, if they do not function appropriately, abnormal growth and development may occur. When a primary manifestation of such abnormal growth was observed to be neoplasia, these protooncogenes were named oncogenes. This nomenclature has persisted despite the ultimate discovery that the unaltered forms of these genes are normal components of the genome.
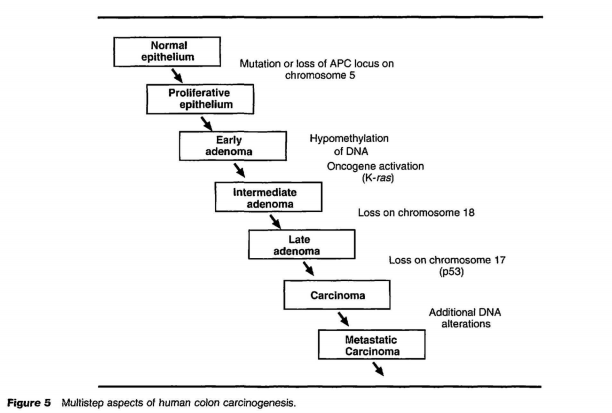
The appearance (phenotype) and function of a tissue is a consequence of which genes are actively producing their programmed product, typically a protein, which in turn affects the structure and function of the cells comprising a given tissue. All somatic cells in the body inherit a complete complement of maternal and paternal genes. The reason that some cells form liver and produce products such as albumin while other cells form kidney tubules that function to excrete substances from the body is a consequence of which genes are expressed in those cells. In liver cells, several critical genes that are important in kidney function are not expressed and vice versa. Specific gene expression and its effect upon tissue phenotype and function are modulated by several intrinsic and extrinsic factors (Figure 6). Since a primary function of many oncogenes is to control cell growth, proliferation, and differentiation, inappropriate expression of these genes would be expected to influence abnormally tissue proliferation and growth. Oncogene activation is a consequence of inappropriate or excessive expression of a protooncogene.
Oncogenes can be activated by several different mechanisms (e.g., retroviral transduction, chromosomal translocation, gene amplification, point mutation, promoter/enhancer insertion, or decreased methylation of promoters). Once activated an oncogene will either be inappropriately expressed (e.g., production of an altered message and protein) or overexpressed (e.g., production of too much of a normal message and protein). Either situation may contribute to the neoplastic process by influencing cellular proliferation and differentiation. Examples of activated or amplified oncogenes detected in human and animal neoplasms are listed in Tables 5 and 6. For some cancers the frequency of oncogene activation is relatively high, while for other cancers the activation of known oncogenes is uncommon. Identification of specific alterations in oncogenes in certain cancers represents a first step in determining the molecular basis of cancer and could eventually lead to the development of molecular intervention and therapeutic strategies. Experimental evidence indicates that oncogene activation can be an early critical event in carcinogenesis, and experimental studies with known chemical carcinogens show that they produce specific alterations in certain oncogenes reflecting the manner in which the carcinogen chemically affects DNA.
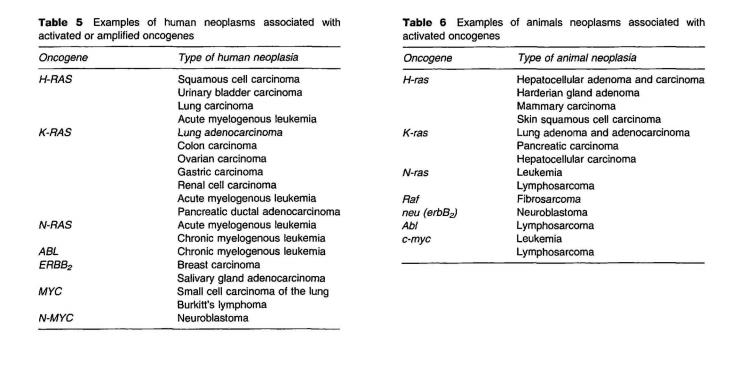
Tumor Suppressor Genes
Tumor suppressor genes, originally called antioncogenes, function to suppress the development of cancerous growth. White oncogenes must be activated to be effective, tumor suppressor genes must be inactivated or lost for cancer to develop. It has been shown that loss or mutation of both paternal and maternal copies, that is, in both alleles, of a tumor suppressor gene must occur to ablate their effect of suppressing cancer formation. A well-known and extensively studied tumor suppressor gene is the retinoblastoma gene [RB-1). In hereditary retinoblastoma an affected child is born with deletions of portions of one allele of chromosome 13 containing the RB-1 gene. If a second event leading to a loss or alteration of the remaining RB-1 allele occurs while retinal cells are undergoing growth during development, the ocular neoplasm, retinoblastoma, frequently present in both eyes, will occur early in life. Loss or alteration of both copies of this tumor suppressor gene is sufficient to cause retinoblastoma. Although named for the disease in which it was discovered, alterations in the RB-1 gene have been detected in breast, lung, prostate, and bone cancers.
Acquisition of Mutations
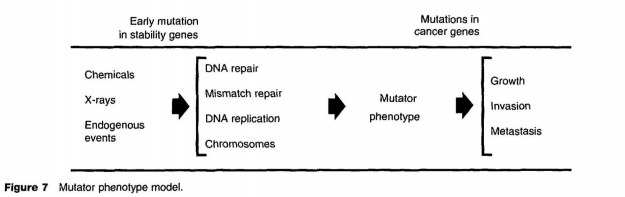
The rate of mutation has been intensely studied in the carcinogenic process. Mutations in cellular DNA can arise during normal cell replication by infidelity in DNA replication (mispairing) as well as by chromosomal deletions, amplifications, or rearrangements. Considering mispairing in nucleotide bases alone, it is estimated that spontaneous mispairing during normal cell replication can occur with a frequency of ~1.4 x 10-10 nucleotide bases per cell division. Since there are ~ 1016 cell divisions per human lifespan and 2 x 109 nucleotide base pairs per genome, a total of 2.8 x 1015 mispairings could occur in a lifetime ((1.4 x 10-10 ) x (2 x 109 ) x 1016). If each mispair led to a mutation that resulted in a cancer, a typical human would have billions of cancers in one average lifetime. Since such estimates of cancer frequency are clearly in excess of what is observed, it is necessary to postulate that events in addition to a single mutation are necessary for most cancers to occur and that many mispairings are repaired or fatal to the cell. There are efficient mechanisms to repair DNA damage, thereby precluding successive accumulation of critical mutations. Cell proliferation is also critical for ‘fixing’ DNA damage since, without production of daughter cells from a damaged mother cell, there would be no inheritance of DNA damage. The cell has relatively efficient mechanisms to repair damage provided there is time prior to cell division. If a tissue is proliferating rapidly, cell division could occur before the cell has time to mend damaged DNA. While all of the above underscore the importance of cell proliferation in carcinogenesis, neoplasia does not occur exclusively or necessarily at higher frequency in tissues that have a rapid intrinsic rate of cell proliferation. Consequently, other important mechanistic factors influence the complex process of carcinogenesis.
In 1994, Loeb et al. proposed that neoplastic cells likely have a higher mutation rate than normal cells (~2 x 10-7 per gene per cell division) and thereby increase the likelihood of neoplastic cells acquiring further mutations conducive to neoplastic growth features. This is referred to as the ‘mutator phenotype’ (Figure 7). It suggests that early mutation in stability genes (i.e., DNA repair, mismatch repair, DNA replication, or chromosome maintenance) will lead to the mutator phenotype and further mutations contribute to the subsequent invasive and metastatic properties of the neoplastic growth. Others argue that the mutation rate is similar between neoplastic and normal cells and that it is the higher rate of cell proliferation in neoplasms that gives them more opportunity to accumulate mutations. The healthy debates continue to feed our quest to prevent and cure the neoplastic process.
Growth Factors, Hormones, and Signal Transduction
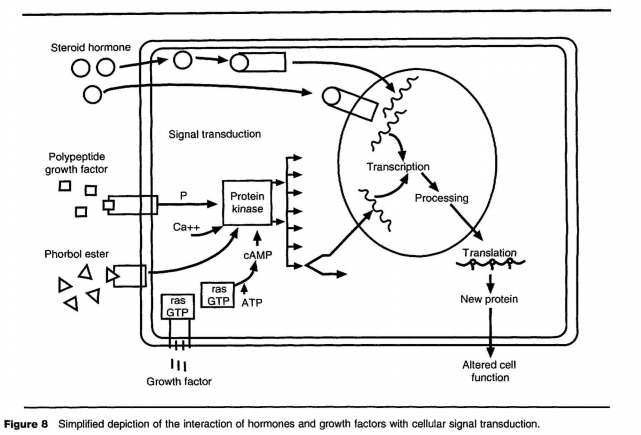
While alterations in cellular DNA are critical in carcinogenesis, some cancer-causing agents, particularly those that are not genotoxic, play a major role in cancer development by indirectly influencing gene expression and growth control by altering signal transduction. While the pivotal role of hormones in the orchestration of tissue growth and development has been appreciated for decades, the recent discovery of polypeptide growth factors has added to our knowledge of the complex constellation of control mechanisms that affect normal cellular growth. Both hormones and growth factors bind to specific cellular receptors and thereby trigger a cascade of intracellular reactions that seem to be associated ultimately with cellular proliferation. This cascade of intracellular reactions is sometimes referred to as signal transduction, the process whereby a stimulus external to the cell triggers a cascade of intracellular biochemical reactions that ultimately lead to expression of specific genes. A simplified depiction of the interaction of hormones and growth factors with cellular signal transduction is presented in Figure 8. This concept is perhaps best exemplified by the process whereby a normal hormone stimulates a tissue to grow. An example is breast development and milk production in response to the hormone prolactin. In this example, prolactin binds to a specific prolactin receptor on the external surface of the cell, which, in turn, triggers a biochemical change inside the cell membrane via molecules that are attached to the external receptor and pass through the cell membrane. This in turn triggers a long chain of biochemical reactions ultimately resulting in a signal to specific genes in the cellular DNA so that they become active. The specific genes, in this example, initiate a program that causes breast cells to divide and secrete milk. The signal transduction pathways in mammalian cells are highly interactive with numerous positive (signal-sending) and negative (signalblocking) feedback loops. An appropriate balance between the positive and negative feedback loops is necessary for the proper functional response to the initial stimulus.
Some forms of cancer development are believed to be facilitated by perturbations in one or more places in the signal transduction pathway. Thus, exposure to certain agents may potentially affect the balance of positive and negative feedback loops in the signal transduction pathway and make cells more susceptible to stimuli that promote growth. An example is the nongenotoxic skin tumor promoter, phorbol ester, which activates protein kinase C, a multifunctional element in the signal transduction pathway that mediates many critical cellular regulatory processes. Treatment of initiated mouse skin with phorbol ester activates protein kinase C, resulting in the development of benign and malignant skin neoplasms. The complexity and pivotal importance of the signal transduction pathways help explain why multiple types of agents influence carcinogenesis, why multiple steps are involved in the carcinogenic process, and why different cancers are so heterogeneous. Signal transduction involves shifts in intracellular ion fluxes for elements such as sodium, potassium, and calcium. It also often involves activation of protein kinase C, an enzyme that phosphorylates many proteins that may be important in producing a mitogenic response. Part of the signal transduction cascade involves increased expression of cyclic adenosine monophosphate, now recognized as a mitogenic signal, and increased expression of one or more cellular protooncogenes. Current research results demonstrate that increasing numbers of protooncogenes and growth factors are integral parts of the signal transduction pathway and, when altered, influence development of cancer by subverting signal transduction.
Telomeres and Telomerase
Telomerase activation appears to be a critical component of the immortalization process in neoplastic cells and it may provide the basis for new therapeutic targets. Telomeres are specialized structures at the ends of chromosomes and telomerase is the enzyme that maintains the length of the telomeres. During each round of cell division there is a loss of a small number of nucleotides causing progressive erosion of genetic material at the end of each chromosome. As the normal cell divides the telomeres shorten and telomerase is inactive. After a certain number of divisions the shortened telomeres signal the cell to cease dividing and the cells become ‘senescent’ or perhaps will die by apoptosis. Germ cells and some neoplastic cells have sustained function of the telomerase enzyme which helps maintain lengthening of the telomeres and promote continued replication. Tumors having an increased telomerase activity suggest a direct effect, but it is only part of the story. For example, p53 is activated by telomerase and in the absence of p53 these cells fail to undergo apoptosis and go on to proliferate.
Heredity and Cancer: Family Cancer Syndromes
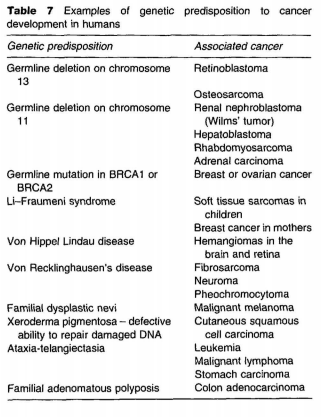
That certain cancers occur in greater frequency within families represents primary empirical evidence for susceptibility based on some hereditary element. Some genetic predispositions exist for cancers of unknown etiology, while interactions between genetic susceptibility and environmental factors are probably responsible for a large proportion of human cancers. Hereditary predispositions include DNA repair deficiencies, inability to detoxify carcinogens, and germline loss or mutations of critical genes. Examples of genetic predispositions to cancer are listed in Table 7 and include neurofibromatosis, retinoblastoma, breast cancer, and adenomatosis of the colon. In many of these instances, one event in the carcinogenic process is believed to be an inherited germline mutation in the DNA. Another inherited anomaly, an inability to repair ultraviolet light-induced DNA damage in individuals with the condition xeroderma pigmentosum, is associated with sensitivity to sunlight and a high incidence of skin neoplasia. However, the majority of genetic damage associated with carcinogenesis is acquired either in utero or from environmental and/or lifestyle factors to which individuals are exposed. Even for those individuals with a hereditary predisposition to neoplasia, additional DNA damage is necessary to lead ultimately to its development. Environmental factors that may increase the risk of cancer development in genetically predisposed individuals include exposure to radiation and agents that stimulate cellular proliferation. Experimental systems in which to study genetic susceptibility to cancer are critically needed to assess the role of gene-environmental interaction in the development of human cancer.
For some cancers in genetically predisposed individuals, the data are consistent with an association between malignant neoplasia and biallelic genetic alteration and this is supported by studies of tumor suppressor genes which prevent the development of neoplasia. Alteration or loss of a single tumor suppressor gene allele is usually insufficient to permit the development of neoplasia. In other words, the remaining functional tumor suppressor gene copy is sufficient to prevent the development of neoplasia; if it is lost or altered, however, neoplasia can develop. This situation occurs in hereditary childhood retinoblastoma, a malignant neoplasm of the retinal cells of the eye. Susceptible individuals inherit a partial loss of one copy (one allele) of chromosome 13, where the retinoblastoma tumor suppressor gene (RB-1) is located, and acquire an alteration or loss of the remaining RB-1 allele during early development. The affected child subsequently develops retinoblastoma, often within the first 2 years of life.
The Immune System and Cancer
The proper functioning of the immune system is evidenced by recovery from common childhood diseases such as mumps and chicken pox. A properly functioning immune system recognizes the foreignness of the agents responsible for these diseases, responds to the infection, eliminates the foreign agents, and confers long-term immunity to subsequent infection by the same or similar agents. It has been proposed that cancer cells are recognized as foreign and that the immune system functions to eliminate such cells from the body before they are transformed into large, malignant neoplasms. This process involves elaboration of antibodies that bind to the cancer cells and activate a process whereby the cancer cells are killed. In addition, specific cells of the immune system, such as cytotoxic T lymphocytes, natural killer cells, and macrophages, have a mechanism for recognizing foreign cells and eliminating them from the body. The process of immune surveillance and removal of cancer cells is facilitated when the cancer cells express surface antigens that are recognized as foreign. Exposure to agents that depress the normal functioning of the immune system can lead indirectly to neoplasia by permitting early persistence and development of recently emergent cancer cells. Once a neoplasm has reached a critical size and growth rate, it may not be possible for even a properly functional immune system to effectively eliminate the neoplastic cells.
Operational Phases and Theoretical Aspects of Carcinogenesis
In addition to being complex, the process of carcinogenesis is typically prolonged, requiring a significant portion of the lifespan to become clinically apparent. While perturbations in cellular DNA are essential to carcinogenesis, they alone are not sufficient to cause cancer in all cases. Thus, in some experimental situations, a few minutes of exposure to a carcinogen is sufficient to result ultimately in cancer, whereas in other situations, exposure to the same carcinogen will nor result in cancer unless there is additional experimental manipulation. Smokers illustrate this principle since many, but not all, ultimately develop lung cancer. In other experimental studies, simultaneous administration of a carcinogen and a second agent may enhance, reduce, or block the carcinogenic process depending on the agent employed. These and other carcinogenesis studies have elucidated some of the mechanisms and factors that influence carcinogenesis, delimited some of the specific stages in the multistep process, and continually reminded us of the complexity of this disease process.
Multistep experimental models of carcinogenesis are useful in defining events in the neoplastic process; provide the foundations for current operational descriptions and hypotheses of the biological mechanisms of carcinogenesis (see Figure 1); are available for many organ systems including the skin, liver, urinary bladder, lung, intestine, mammary gland, and pancreas; and frequently are derived from studies of the effects of chemical agents on laboratory animals. The operational phases of carcinogenesis include initiation, promotion, and progression.
Initiation
During the initiation phase of chemical carcinogenesis, a chemical agent or carcinogen interacts with a cell to produce an irreversible change that may ultimately be manifested by a capacity for autonomous growth. The initiated cell appears normal, and the capacity for autonomous growth may remain latent for weeks, months, or years. Initiation implies alteration of the affected cell’s DNA at one or more sites, a mutational event that is by definition hereditary. Direct-acting carcinogens interact directly with cellular DNA to produce the damage while indirect-acting carcinogens must be metabolized by the cell to produce a chemical species that interacts with cellular DNA. The majority of damaged cells have the ability to repair the damaged DNA over a period of days or weeks; however, if a cell undergoes cell division with its attendant DNA replication prior to repair of the DNA damage, the DNA alteration becomes ‘fixed’, is no longer reparable, and is inherited by all subsequent daughter cells. The operational phase of initiation is relatively short and may occur within hours or days. In contrast, the progression of an initiated cell to a fully malignant neoplasm is a prolonged process requiring months in animals and years in humans. Based on a large body of evidence that most initiators are mutagenic or genotoxic, a battery of short-term mutagenicity tests in bacteria and cell culture systems has evolved to identify chemicals with genotoxic properties. Once identified, such chemicals should be rigorously regulated to prevent human exposure. This approach is considered prudent because of the irreversible and hereditary nature of the changes that occur during initiation. Indeed, it is generally believed that even a single molecule of a mutagenic substance is potentially sufficient to damage DNA irreversibly. Thus, for practical purposes there is no threshold or safe level of exposure to a mutagenic agent. Salient features of initiation are listed in Table 8.
Initiators interact with host cellular macromolecules and nucleic acids in specific patterns. The majority of known initiators have both initiating and promoting (see below) activity and can thus induce neoplasms rapidly and in high yield when there is repeated or high-level exposure. When given at sufficiently low single doses, an initiated cell requires subsequent promotion for the development of any neoplasia. Thus, the dose of an initiator is a critical determinant of its carcinogenic potential.

Promotion
Promotion is classically considered that portion of the multistep carcinogenic process in which specific agents, known as promoters, enhance the development of neoplasms by providing initiated cells with a selective growth advantage over the surrounding normal cells. The characteristic features of promotion are listed in Table 8. By definition, a promoter is given at some time after chemically induced or fortuitous initiation and the experimental doses of promoting agent are insufficient to produce cancer without prior initiation. When classical promoters are administered at sufficiently high doses and for prolonged intervals, neoplasia can occur without evidence of prior initiation. Under these conditions, a promoting agent must be considered a complete carcinogen unless fortuitous initiation from background radiation, dietary contaminants, environmental toxins, etc., is believed to have occurred. However, under experimental conditions commonly employed in short- and medium-term initiation-promotion experiments, neoplasia does not typically occur in animals that are not previously initiated.
The temporal sequence of promoter administration is critical to the operational definition of promotion. The agent must be administered after initiation and cause enhancement of the neoplastic process to be considered a promoter. If an agent is given simultaneously with an initiator and results in enhancement of development of neoplasms, it is regarded as a cocarcinogen rather than a promoter. While some promoters are cocarcinogenic (e.g., phorbol esters), not all promoters (e.g., phenobarbital and phenol) possess cocarcinogenicity and, conversely, not all cocarcinogens are promoters. Under these same conditions of simultaneous administration, a diminution in the neoplasm response is considered evidence of anticarcinogenic activity. Several rodent liver tumor promoters, which are active when administered after a variety of initiators, prevent or delay the development of liver neoplasms when added to diets along with an active carcinogen. Finally, reversing the order of administration by giving a known promoter prior to an initiator may prevent the expression of carcinogenic activity on the part of the initiator.
While upper and lower thresholds have been demonstrated experimentally for promoters, some consider that, in an absolute sense, it is statistically impossible to prove or disprove the existence of thresholds for promoters for much the same reasons that this cannot be done for initiators. One can never be certain that an apparent no-effect level would, indeed, be without effect if a sufficiently large enough number of animals were used. Promoters include agents such as drugs, plant products, and hormones that do not directly interact with host cellular DNA (are not genotoxic) but somehow influence the expression of genetic information encoded in the cellular DNA. Experimental evidence suggests that regulation of gene expression is unique to the nature of the promoting agent administered. Some promoters are believed to produce their effect by interaction with receptors in the cell membrane, cytoplasm, or nucleus (e.g., hormones, dioxin, phorbol ester, and polychlorinated biphenyls). Alternatively, promoting agents may exert their effect through their molecular orientation at cellular interfaces. Other promoters may selectively stimulate DNA synthesis and enhance cell proliferation in initiated cells, thereby giving them a selective growth advantage over surrounding normal cells.
Promoters appear to have a relatively high tissue specificity. Thus, phenobarbital functions as a promoter for rodent liver neoplasia but not urinary bladder neoplasia. Saccharin, on the other hand, promotes urinary bladder neoplasia but not liver neoplasia in the rat. Similarly, 12-o-tetradecanoylphorbol-13-acetate (phorbol ester) is a potent skin and forestomach neoplasm promoter in the laboratory rodent but has no appreciable activity in the liver. Other agents, such as the antioxidants 3-t-butyl-4-methoxyphenol and 2,6-di-t-butyl-4-methoxyphenol, may act as promoters in one organ and antipromoters in another and have no effect in a third organ. Thus, the practical definition of a promoter must include the designation of the susceptible tissue.
Tumor promotion may be modulated by several factors such as age, sex, diet, and hormone balance. The correlation of increased rates of breast cancer in women following a ‘Western’ lifestyle has implicated meat and fat consumption as playing an important role in breast cancer development. Experimental demonstration of the role of a high-fat diet in the promotion of mammary cancer in rats exposed to the mammary carcinogen dimethylbenzanthracene has been documented. Similarly, bile acids, as modulated by fat consumption, are known promoters of rat liver carcinogenesis and human colorectal cancer. Ageand sex-associated modulations in hormonal levels of estrogens, progesterone, and androgens have been implicated as potential promoters of breast cancer on the basis of epidemiological studies in humans. Experimental studies have repeatedly shown that these hormones, in addition to pituitary prolactin, serve to promote mammary cancer in rats initiated with mammary carcinogens.
Progression
Progression is that part of the multistep neoplastic process associated with the development of an initiated cell into a biologically malignant cell population. In common usage progression is frequently used to signify the stages whereby a benign proliferation becomes malignant or, alternatively, where a neoplasm develops from a low grade to a high grade of malignancy. During progression neoplasms show increased invasiveness; develop the ability to metastasize; and then biochemical, metabolic, and morphologic characteristics are altered.
Expression of tumor cell heterogeneity, an important characteristic of tumor progression, includes production of antigenic and protein product variants, ability to elaborate angiogenesis factors, emergence of chromosomal variants, development of metastatic capability, alterations in metabolism, and a decrease in sensitivity to radiation. The development of intra neoplastic diversity may result from increasing genetic damage. Alternatively, the heterogeneity observed in tumor progression may be generated by epigenetic, regulatory mechanisms that are a part of the process of promotion. More than likely, genetic and nongenetic events subsequent to initiation operate in a nonmutually exclusive manner during progression, possibly in an ordered cascade of latter events superimposed upon earlier events.
The most plausible mechanism of progression invokes the notion that, during the process of tumor growth, there is a selection that favors enhanced growth of a subpopulation of the neoplastic cells. In support of this mechanism is increased phenotypic heterogeneity observed in malignant but not benign neoplastic proliferations. Presumably, a variety of subpopulations arises, and it is only a matter of time before the emergence of a subpopulation with more malignant biological characteristics or at least an accelerated growth advantage. This can be observed occasionally during experimental hepatocarcinogenesis when a phenotypically distinguishable carcinoma can be observed arising within an existing adenoma.
Distinction between tumor promotion and tumor progression is not readily discernible in the routine histopathologic evaluation of neoplasms and may be somewhat academic because promotion may be considered part of the process of progression. In both situations the critical event is accentuated growth. What is believed to distinguish progression from promotion is the presence of structural genomic alterations in the former and their absence in the latter. Both structural genomic changes and biochemical changes associated with tumor progression cannot be defined by conventional histopathology. Established and emerging technologies centered around histochemistry, immunocytochemistry, in situ hybridization, identification of activated oncogenes, loss of tumor suppressor genes, gene expression, proteomic and metabolomic profiling and discovery offer promise to distinguish various stages of progression in the evolution from benign to malignant neoplasms.
Exogenous Factors Influencing Carcinogenesis
Important exogenous factors that contribute to induction of cancer include natural and synthetic chemicals, environmental exposures to ultraviolet and medical radiation, diet and lifestyle, and infectious agents, such as viruses, parasites, and bacteria. Evidence for a causal association between exogenous factors and neoplasia is derived from studies of epidemiology, occupationally common cancers, and animal models.
Chemical and Physical Agents and Lifestyle Factors
Many chemicals that cause cancer interact directly with and alter DNA or are metabolized to chemical derivatives capable of doing so. Exposure to carcinogens can occur in certain occupational settings. Associations of human hepatic angiosarcomas with workplace exposure to vinyl chloride, pulmonary mesotheliomas with exposure to asbestos fibers, and leukemia with benzene are well-known examples. Exposure to other carcinogenic agents may occur in the diet or as a consequence of certain lifestyle practices, such as cigarette smoking associated with pulmonary cancer and high animal fat diets linked to breast and colon cancer. Strong associations have been made between exposure of light-skinned individuals to ultraviolet radiation and skin cancer. Exposure to occupational ionizing radiation, X-rays, and medical use of radioisotopes has also been associated with human neoplasia. Examples include leukemias in radiologists and atom bomb victims, lung cancer in uranium mineworkers, and thyroid and breast cancer following diagnostic or therapeutic use of radiation.
Infectious Agents and Inflammation
Viral, parasitic, and bacterial infections have been linked to cancer (Table 9). DNA viruses such as Epstein-Barr, hepatitis B, hepatitis C, papillomaviruses, and Kaposi sarcoma herpes virus and RNA viruses such as human T-cell leukemia virus type I and human immunodeficiency virus have been implicated in causing cancer in humans and are listed as ‘known-to-cause-cancer’ in humans by the International Agency for Research on Cancer (1ARC). In man, the liver fluke, Opistborchis viverrini, is associated with the development of cholangiocarcinomas of the liver and the blood fluke, Schistosoma haematobium, with carcinoma of the urinary bladder. There is evidence that chronic Helicobacter pylori infection of the stomach in man is not only related to gastrointestinal ulcers, but also may be linked to gastric carcinoma or lymphoma development.
For oncogenic viruses, the viral or host genes generally drive the neoplastic process while with some agents there appears to be an association of chronic inflammation and nitric oxide (NO) production in the development of cancer. When DNA viruses infect cells, the viral DNA inserts itself wholly or partially into the genome of the infected cell. It appears that such integration of viral DNA into the mammalian genome is sometimes sufficient to cause neoplastic transformation of the infected cell, which is accompanied by the production of new proteins essential for the neoplastic process. RNA viruses associated with neoplasia are chiefly represented by the retroviruses. RNA viruses possess an enzyme called reverse transcriptase, which is capable of forming a DNA copy of the viral RNA when the virus infects a host cell. This DNA ultimately inserts itself into the host genome in much the same way as DNA viruses do, possibly resulting in the development of neoplasia.
The role of inflammation in cancer development is being intensely studied. There are a number of chronic inflammatory conditions, infectious and noninfectious, in man and animals associated with an increasing risk of cancer and there are many investigators examining the role of NO and oxygen radical damage to DNA or other cellular processes such as cell proliferation and apoptosis. NO induces p53, prevents apoptosis in cells such as endothelium, promotes angiogenesis, and inhibits DNA-repair activities – all processes that might provide a selective advantage to neoplastic cell growth.

Identification of Carcinogenic Agents
There are two methods utilized to identify potential human carcinogens, the most direct of which is based on retrospective epidemiological studies in human populations using existing historical records associated with known cases of neoplasia. These records include death certificates where cause of death is indicated; hospital records; responses to questionnaires that document environmental or work-associated exposure to potential carcinogenic agents; and studies of neoplasia in culturally, ethnically, or religiously distinctive human populations. Association of cigarette smoking with lung cancer and exposure to asbestos with mesotheliomas was the result of such retrospective epidemiological work. Prospective epidemiological studies identify a given population of individuals who agree to be monitored for several years to permit identification of potential carcinogenic factors associated with neoplasms which may occur.
Another method used to identify potential human carcinogens involves testing known chemicals and agents in experimental animals. Such tests have been referred to as animal bioassays and are typically conducted using rats and mice exposed to high doses of the suspect agent for a large portion (typically 2 years) of their lifespan. If such agents are observed to produce neoplasia in the experimental animals, the agent is regarded as a potential human carcinogen. In countries throughout the world, legal requirements mandate that all new chemical agents and drugs be tested in animal bioassays to determine whether they cause cancer in the test animals. Additionally, since the mid-1960s in the United States, the National Cancer Institute and currently the National Toxicology Program have collectively conducted animal bioassays on more than 500 chemical agents to assess their potential to cause cancer.
Interpretation of results from human epidemiological studies and animal bioassays to identify carcinogenic agents has often proved difficult and controversial. Humans are rarely exposed to only one potential cancer-causing agent in their lifetime, and the amount and duration of that exposure may be difficult or impossible to quantify rigorously. Many years may intervene between exposure to a potential carcinogen and ultimate development of neoplasia, making accurate assessment of cause and effect almost impossible. Despite such limitations, epidemiological studies that clearly show an association between a given chemical exposure or lifestyle habit with an enhanced rate of a specific cancer are regarded as the most relevant method for identification of human carcinogens. While animal bioassays have proved useful for the identification of agents that can cause cancer in the laboratory rodent, they only identify an agent as potentially hazardous to human health. Additional facts and factors must be considered in classifying such an agent as a likely human carcinogen.
The current approach for assessing the scientific relevance of either epidemiological or animal bioassay results to human health risk involves a ‘weight-of-evidence’ procedure in which national and international panels of expert scientists from several disciplines examine all available information on the suspect agent in making their assessment. Included in this analysis are the strength of the epidemiological evidence, the dose-response curve of the animal response, comparative species metabolism and ability to extrapolate between species, likely mechanism of cancer induction for the agent in question, the genotoxicity of the agent, the amount of the agent in the environment, and the number of people potentially exposed to the agent. Based on this type of analysis, so far 88 agents have been classified as known human carcinogens by the International Agency for Research on Cancer (some of which are in Table 10) and 64 more agents have been designated as probable human carcinogens. The 10th US Health and Human Services Annual Report on Carcinogens lists 49 known human carcinogens and 174 substances that are reasonably anticipated to be human carcinogens.
Molecular Epidemiology of Cancer
The molecular epidemiology of cancer is the study of molecular alterations, primarily mutations, in investigating the causative agents of cancer as well as identifying individual cancer risk. The possibility of identifying cancer-causing agents based on the occurrence of predictable molecular alterations that are found in the neoplasm is intriguing. It is based on the hypothesis that there are carcinogen-specific patterns of mutations that reflect direct interactions of carcinogens with cancer genes. For example, lung and colon cancers from people who smoke rend to have a specific mutation in the ras oncogene or p53 tumor suppressor gene (i.e., mostly a G-T nucleotide base substitution) and that this mutation is likely due to the direct interaction of the carcinogen in smoke benzo(a)pyrene with DNA. Such chemical-specific mutational profiles (or ‘molecular signatures’) have been used to support a causal association between particular genetic events in tumors and a specific carcinogen such as neoplasms associated with exposure to radon, aflatoxin B1, vinyl chloride, and the nitrosamines (Tables 11 and 12). The strongest evidence for linkage between a cancer-causing agent and a specific type of neoplasm is that of the CC-TT double base changes observed in skin neoplasms of man and animals. This mutation is consistent with the predicted UV-induced damage of dipyrimidine dimers. In liver tumors from persons living in geographic areas with a high exposure to aflatoxin B1) there is a frequent mutation at the third nucleotide pair of codon 249 in the p53 gene, suggesting the mutation is chemical-specific and imparts a specific growth or survival advantage to the mutated liver cells.
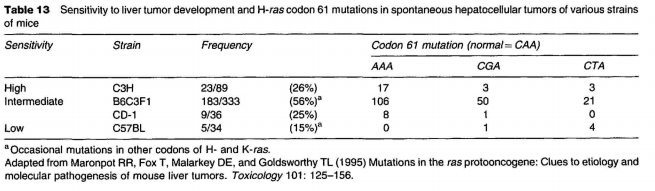
Animal studies have confirmed that there are certainly chemical-specific mutational profiles in neoplasms; however, there are many examples where the mutational profile varies by strain (Table 13), species, dose, or dosing regiment. For example, diethylnitrosamine, a strong, cross-species hepatocarcinogen, will induce liver neoplasms in mice, rats, and rainbow trout, but the frequency and type of ras mutation in the neoplasm varies widely, and the mutations are not simply a reflection of direct DNA interaction (Table 14). In some studies, in vitro mutation assays were poor predictors of liver tumor mutation profiles in the mouse. In this complex process, carcinogens might also be influencing events such as DNA repair, oxidative DNA damage, methylation, cell death, proliferation, and/or a hypermutable state.
Molecular epidemiologic studies aimed at identifying an individual’s risk of developing cancer have found that persons with germline mutations in cancer genes (i.e., BRCA1 or BRCA2) or variations (polymorphisms) of carcinogen-metabolizing enzyme activities (i.e., cytochrome P-450s or glutathioneS-transferases) or DNA repair capacities can be at increased risk of developing neoplasia in their lifetime. High-throughput analyses to examine single nucleotide polymorphisms (SNPs) are being used to search for biomarkers of cancer risk in individuals and some of this information is being used to help people take preventive measures to decrease their risk of developing cancer.


Summary and Conclusions
All of life is a balancing act of good versus evil and production versus destruction. Similar balancing factors are evident in carcinogenesis where regulatory mechanisms for tissue proliferation are balanced against those for cellular differentiation. It is well established that carcinogenesis requires the accumulation of multiple alterations in the genome of the affected (cancer) cells. At the genetic level, two opposing classes of genes, oncogenes, and tumor suppressor genes, have been implicated in the carcinogenic process. In addition, the development of cancer is influenced by host factors such as age, sex, diet, nutrition, general health status, and inherited predispositions for cancer and by complex positive and negative intracellular signaling mechanisms. Treatment of cancer is based on our understanding of the mechanistic underpinnings of the carcinogenic process and attempts to shift the balance of critical factors in favor of patient survival. The probability of developing cancer is directly proportional to the intensity, route, and duration of exposure to cancer causing factors as well as genetic susceptibility. Public health strategies are based on the premise that reduction or prevention of exposure to cancer-causing factors will decrease the incidence of cancer.
Further Reading
Barrett JC (ed.) (1987) Mechanisms of Environmental Carcinogenesis, vols. I and II. Boca Raton, FL: CRC Press.
Hussain SP, Hofseth LJ, and Harris CC (2001) Tumor suppressor genes: At the crossroads of molecular carcinogenesis, molecular epidemiology and human risk assessment. Lung Cancer 34: S7-S15.
Klein-Szanto JP, Anderson MW, Barrett JC, and Slaga TJ (eds.) (1992) Comparative Molecular Carcinogenesis. New York: Wiley-l.iss.
Loeb LA (1994) Microsatellite instability marker of a mutator phenotype in cancer. Cancer Research 54: 5059- 5063.
Marx J (2002) Debate surges over the origins of genomic defects in cancer. Science 297: 544-546.
Mastorides S and Maronpot RR (2002) Carcinogenesis. In: Haschek W, Rousseaux CG, and Wallig MA (eds.) Handbook of Toxicologic Pathology, 2nd edn., pp. 83-122. New York: Academic Press.
Mendelsohn J, Howley PM, Israel MA, and Liorta LA (eds.) (2001) The Molecular Basis of Cancer, 2nd edn. Philadelphia: Saunders.
Pitot HC (1986) Fundamentals of Oncology, 3rd edn. New York: Dekker.
Tannock IF and Hill RP (1987) The Basic Science of Oncology. New York: Pergamon.
Vogelstein B and Kinzler KW (2004) Cancer genes and the pathways they control. Nature Medicine 10(8): 789-799.
Glossary
Adenomatosis
a condition in which numerous adenomatous growths develop in a tissue.
Allele
one of the two gene pairs situated at the same location on a chromosome; one allele is inherited from the mother and the other from the father; characteristics such as being short or tall or having blue eyes versus brown eyes are determined by the expression of inherited alleles.
Anaplasia
lack of normal organizational or structural differentiation of a tissue; anaplastic cells are typically poorly differentiated.
Angiogenesis
the development of blood vessels.
Benign
a classification of anticipated biological behavior of neoplasms in which the prognosis for survival is good; benign neoplasms grow slowly, remain localized, and usually cause little harm to the host.
Biallelic damage
damage to both maternal and paternal copies of a gene.
Cancer
generally refers to a malignant neoplasm.
Carcinogenesis
the process of development of cancer or neoplasms.
Choristoma
a mass or collection of well-differentiated cells from one organ found within another organ, for example, adrenal tissue present in the lung.
Clonal
pertaining to a clone; a line of cells descended from a single cell,
Cocarcinogen
an agent that has no inherent carcinogenic activity by itself hut is capable of augmenting neoplasm formation when given simultaneously with a genotoxic carcinogen.
DNA
abbreviation for deoxyribonucleic acid; the basic building block of genetic material in all organisms except RNA viruses.
Dysplasia
disordered tissue formation characterized by changes in size, shape, and orientational relationships of adult types of cells; primarily seen in epithelial cells.
Epithelial cell
cells which line the internal and external surfaces of the body and form the bulk of many of the major organs of the body; they are formed from the embryonic germ layers known as entoderm and ectoderm.
Gene
the basic biological unit of heredity which is located on a chromosome.
Genome
the total complement of genes present in the set of chromosomes characteristic of a given organism.
Genotoxic
toxic to DNA; an agent or process that interacts with cellular DNA either directly or after metabolic transformation; mutagens are genotoxic agents.
Grade (grading)
a subjective evaluation of the morphologic characteristics of a neoplasm based on the degree of anaplasia and proliferation evident from microscopic examination as a measure of biological outcome or degree of malignancy.
Growth factors
agents that contribute to and stimulate tissue growth.
Hamartoma
a localized overgrowth of differentiated cells that have an altered growth pattern in relation to the tissue in which they are found, for example, a nodule of disorganized striated muscle fibers within a normal skeletal muscle.
Hepatocarcinogenesis
the development of liver cancer.
Heterozygous
having different alleles at a specific position on a chromosome.
Histogenetic
pertaining to the origin, formation, or development of tissues from undifferentiated embryonic germ cell layers.
Homeostatic
pertaining to the natural state of equilibrium of the normal internal environment of the body; maintained by complex positive and negative feedback control mechanisms.
Homozygous
having identical alleles at a specific position on a chromosome.
Hyperplasia
a numerical increase in the number of normal-appearing cells within a tissue or organ.
Immune system
a primary defense system in the body capable of attacking and potentially destroying cancer cells; consists of lymphoid and related tissues from which cells are recruited to produce antibodies or to directly attack cancer cells.
Initiation
the first operational phase of the process of carcinogenesis during which a cell sustains a heritable alteration in DNA.
Malignant
a classification of anticipated biological behavior of neoplasms in which the prognosis for survival is poor; malignant neoplasms grow rapidly, invade, destroy tissue, and are usually fatal.
Mesenchymal cell
cells derived from embryonic mesoderm, which constitute the supporting structure of tissue such as connective tissue, blood vessels, muscles, and bones.
Metaplasia
the substitution of one type of fully differentiated cells for the fully differentiated cell type normally present in a given tissue.
Metastasize
the spreading of neoplastic cells from a primary site of origin to a distant, noncontiguous site where their growth occurs.
Mitogenic
stimulating cell proliferation or division; causes mitosis.
Mutation
a structural alteration in DNA that is hereditary and may give rise to an altered phenotype.
Neoplasia
the process of the development of neoplasms; essentially synonymous with carcinogenesis.
Neoplasm
new and typically abnormal growth which is generally uncontrolled and becomes progressively more serious with time.
Neurofibromatosis
a hereditary condition of the nervous system and other tissues of the body characterized by development of numerous neoplasms (neurofibromas) distributed over the entire body.
Nucleotide
a biochemical component of DNA that consists of a purine or pyrimidine base, a ribose or deoxyribose sugar, and a phosphate group; a basic building block of DNA.
Oncogene
a so-called cancer gene because alterations in its structure or expression are typically associated with neoplasms; an activated form of a protooncogene.
Oncogene activation
the process whereby a protooncogene is altered such that it stimulates enhanced cellular growth; several different mechanisms can lead to such activation.
Oncology
the study of neoplasia or carcinogenesis.
Phenotype
the physical appearance, biochemical makeup, and physiological behavior of an individual.
Preneoplasia
refers to the recognizable structural changes in a tissue that are sometimes antecedent to the development of neoplasia; the presence of preneoplasia indicates an increased probability for the development of neoplasia.
Progression
an operational phase of carcinogenesis associated with the development of an initiated cell into a fully malignant neoplasm; sometimes used in a more limited sense to refer to the change from a benign neoplasm to a fully malignant neoplasm.
Promotion
an operational phase of carcinogenesis in which there is enhancement of neoplasm formation when an agent (the promoter) is administered after exposure to a genotoxic carcinogen.
Protooncogene
a normal cellular structural gene that, when activated by mutations, amplifications, rearrangements, or viral transduction, functions as an oncogene and is associated with neoplasia; regulates normal processes related to cell growth and differentiation.
Retinoblastoma
an ocular neoplasm arising from germ cells of the retina.
Retroviruses
a large group of RNA viruses.
Stage (staging)
a subjective assessment of the extent to which a neoplasm has spread in the body and, thus, an indication of the patient’s clinical prognosis.
Threshold
the level of an agent below which no physiological, biochemical, or pathological effect can be measured.
Tumor
any tissue enlargement or swelling; frequently used as equivalent to a benign neoplasm.
Tumor suppressor gene
a gene that normally functions to suppress uncontrolled tissue growth.
Weight of evidence
an approach for assessing the potential carcinogenic risk of an agent by considering all available information relative to the biological action of the agent.