Perillaldehyde, a natural monocyclic terpenoid found most abundantly in the herb perilla, has a long history of use as a flavouring ingredient to add spiciness and citrus taste to foods. Previously, it was judged to be safe by several international expert panels. To confirm the safety of flavourings placed on the European Union list of flavourings, perillaldehyde was selected by the European Food Safety Authority as a representative of a subgroup of alicyclic aldehyde flavouring substances to be evaluated for genotoxic potential. Perillaldehyde was tested in a bacterial reverse mutation assay, an in vitro micronucleus assay in human lymphocytes, an HPRT assay in mouse lymphoma cells, and a micronucleus/comet assay in Han Wistar rats. In contrast to previously published results, perillaldehyde induced mutation in Salmonella typhimurium strain TA98 in the absence of metabolic activation. The comet assay was negative for duodenum and weakly positive for liver but only at a hepatotoxic dose of perillaldehyde. All other genotoxicity assays were negative. These data do not provide an indication of any genotoxic potential for perillaldehyde, and they provide the primary basis for recent scientific opinions regarding perillaldehyde genotoxicity announced by several international organizations responsible for safety assessment of food additives and flavourings.
Key Words: genotoxicity, flavouring agent, perillaldehyde, p-mentha-1,8-dien-7-al, perilla aldehyde, DNA damage
Abbreviations: EFSA, European Food Safety Authority; FAO, Food and Agriculture Organization of the United Nations; FEMA, Flavor and Extract Manufacturers Association; GLP, Good Laboratory Practice; HPRT, hypoxanthine-guanine phosphoribosyl transferase; ICH, International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; JaCVAM, Japanese Center for the Validation of Alternative Methods; JECFA, FAO/WHO Joint Expert Committee on Food Additives; ln, natural log; MN, micronucleus or micronuclei; MN-PCE, micronucleated polychromatic erythrocytes(s); OECD, Organization for Economic Cooperation and Development; PCE, polychromatic erythrocytes; (Q)SAR, quantitative structure activity relationship; UKEMS, United Kingdom Environmental Mutagen Society; WHO, World Health Organization
1. Introduction
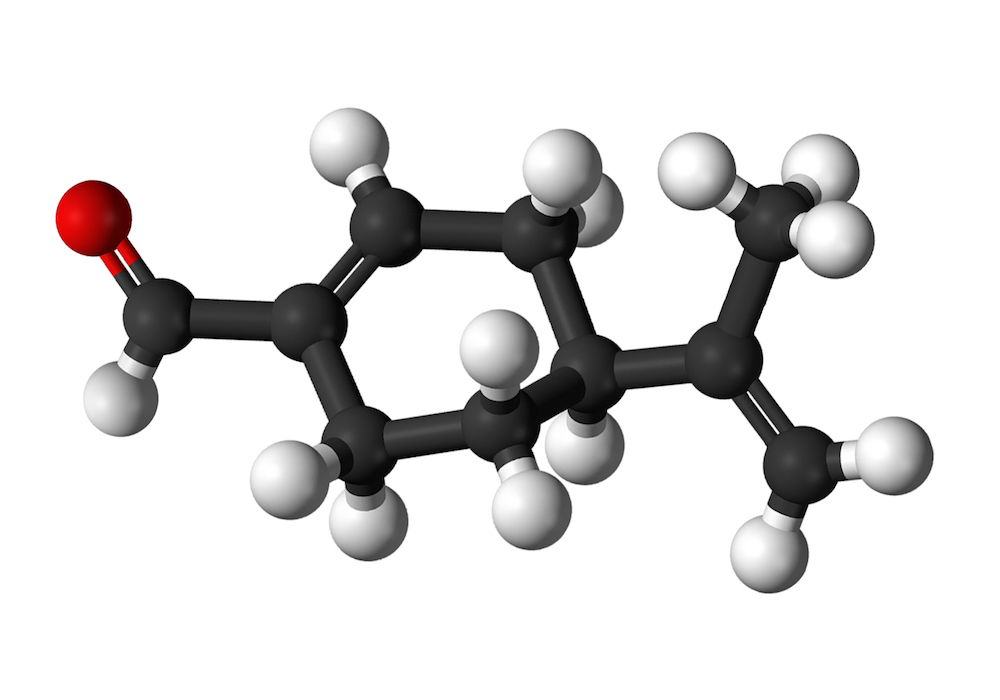
Perillaldehyde (also known as l-perillaldehyde, perilla aldehyde, l-perilla aldehyde, and p- mentha-1,8-dien-7-al) is a natural compound found abundantly in the annual herb perilla and the peel of citrus fruits. Perillaldehyde and volatile oils from perilla rich in perillaldehyde are used as flavouring agents to add spiciness and a woody, citrus taste to foods such as baked goods, puddings, meat products, salad dressing, sauces, pickled vegetables, and beverages. It is also used for its mint-like cinnamon odor in the perfume industry and is being investigated for potential hypolipidemic, anti-inflammatory, neuroprotective, antidepressant-like, and anti-fungal effects (Ji et al., 2014; Omari-Siaw et al., 2016; Tian et al., 2015; Xu et al., 2014). Perillaldehyde is considered as “generally recognized as safe” (GRAS) by the Expert Panel of the U.S. Flavor and Extract Manufacturers Association (FEMA) (Oser and Ford, 1978), was judged to be safe by the Food and Agriculture Organization of the United Nations (FAO)/World Health Organization (WHO) Joint Expert Committee on Food Additives (JECFA) (JECFA, 2003), and has been designated unlikely to harm human health by the Japanese Ministry of Health, Labour and Welfare under the Food Sanitation Act (http://www.mhlw.go.jp/english/topics/foodsafety/foodadditives/index.html) since 1948.
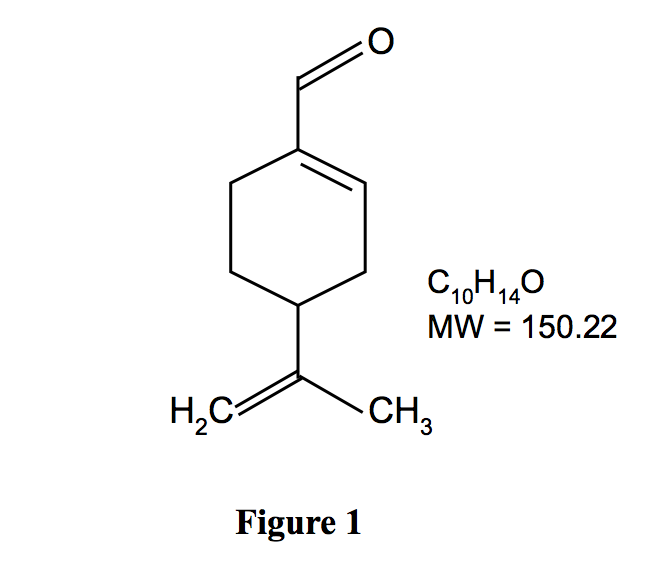
Perillaldehyde is a monocyclic terpenoid containing an α,-unsaturated aldehyde functional group (Fig. 1). It is quickly metabolized, largely by oxidation of the side chain to a carboxylic acid, which is excreted unchanged and as conjugates (JECFA, 2003). In a safety assessment program for confirming the safety of flavourings listed on the European Union (EU) list, the European Food Safety Authority (EFSA) Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids requested additional data related to the possible genotoxic potential of flavouring substances containing α,β-unsaturated aldehyde and ketone structures, or their potential precursors
In response to the request of EFSA for genotoxicity data for use in risk assessment, perillaldehyde was evaluated in a Good Laboratory Practice (GLP)-test battery compliant with EFSA and OECD guidance on genotoxicity testing (EFSA, 2008a, 2012; OECD, 1997a, b, c, 2008). Specifically, perillaldehyde was evaluated in a bacterial reverse mutation assay (OECD 471) using Salmonella and E. coli tester strains, an in vitro MN assay using human peripheral blood lymphocytes (OECD 487), and an in vitro HPRT mutation assay in mouse L5178Y lymphoma cells (OECD 476). In addition, a combined MN (OECD 474) and comet assay was conducted in male Han Wistar rats. Although an OECD test guideline for the in vivo comet assay did not exist at the time this study was conducted, the assay was performed in accordance with recommendations of EFSA and expert working groups (Burlinson et al., 2007; EFSA, 2012; Tice et al., 2000). The results of this comprehensive genotoxicity testing of perillaldehyde are reported.
2. Material and methods
2.1. Chemicals
All genotoxicity assays were conducted according to OECD test guidelines or EFSA guidance (comet assay) and were GLP-compliant with the exception that dose formulations were not analyzed for achieved concentration. Perillaldehyde (91.9-94.2% pure; CAS No. 2111-75-3; Nippon Terpene Chemicals, Inc., Kobe, Japan) solutions were prepared by dissolving in anhydrous analytical grade dimethyl sulfoxide (DMSO) or suspending in corn oil under subdued light conditions with continual stirring before and during dosing. The test article solutions were protected from light and used within 2 (MN/comet and mouse lymphoma assays) or 5 (bacterial mutagenicity and in vitro MN assays) hours of initial formulation. Sodium azide, mitomycin C, vinblastine, and ethyl methanesulfonate were formulated in water; 2-nitrofluorene, 9- aminoacridine, benzo[a]pyrene, 2-aminoanthracene, and cyclophosphamide were formulated in DMSO. Aliquots of stock solutions were either prepared in advance and stored refrigerated (benzo[a]pyrene and mitomycin C for the bacterial reverse mutation test) or frozen at -80°C (2- nitrofluorene, 9-aminoacridine, sodium azide, and cyclophosphamide) in the dark, or they were prepared immediately prior to use (ethyl methanesulfonate, vinblastine, and mitomycin C for the in vitro MN assay). All chemicals and reagents were obtained from Sigma-Aldrich Chemical Co. (Poole, UK) or equivalent suppliers unless specifically stated otherwise.
2.2. Bacterial reverse mutation assay
The procedures used in this study were in accordance with OECD Guideline 471 (OECD, 1997a). Mutagenicity assays of perillaldehyde, with and without metabolic activation, were conducted using the following five strains of Salmonella typhimurium bacteria (TA98, TA100, TA1535, TA1537 and TA102). All the tester strains, with the exception of strain TA102, were originally obtained from the UK NCTC. Strain TA102 was derived from a culture obtained from Glaxo Group Research Limited. All strains were checked previously for the maintenance of genetic markers. Metabolic activation was provided by 10% liver post-mitochondrial fraction (S9) prepared from male Sprague Dawley rats induced with Aroclor 1254 (Moltox, Boone, NC). The composition of the S9 mix was: 10% S9, 8 mM MgCl2, 33 mM KCl, 1.5 mg/mL glucose-6- phosphate, 3.2 mg/mL -nicotinamide adenine dinucleotide phosphate (NADP), 0.1 M phosphate buffer, 40 μg/mL L-histidine HCl (in 250 mM MgCl2), and 49 μg/mL d-biotin. Strain specific positive controls tested without metabolic activation were 2-nitrofluorene (TA98; 5 μg/plate), sodium azide (TA100 and TA1535; 2 μg/plate), 9-aminoacridine (TA1537; 50 μg/plate), and mitomycin C (TA102; 0.2 μg/plate). Benzo[a]pyrene (10 μg/plate) and 2- aminoanthracene (TA100, TA1535, TA1537 at 5 μg/plate; TA102 at 20 μg/plate) were used as the positive controls for TA98, and all other strains, respectively, tested with metabolic activation. Bacteria were cultured at 37°C for 10 hours in nutrient broth containing ampicillin (TA98, TA100) or ampicillin and tetracycline (TA102) as appropriate. Incubation was carried out with shaking in an anhydric incubator. All treatments were completed within 6 hours of the end of the incubation period. For plate incorporation assays, bacteria, control or test article formulation, and 10% S9 mix or buffer were added to molten agar at 46±1°C, mixed rapidly, and poured onto Vogel-Bonner E plates. For the pre-incubation assay, perillaldehyde or control formulation, bacteria, and 10% S9 mix were mixed and incubated at 37±1°C for 1 hour prior to the addition of molten agar and plating. Once set, triplicate plates per concentration (five plates for vehicle control) were incubated at 37°C in the dark for 3 days. Colonies were counted using the Sorcerer Colony Counter (Perceptive Instruments, Ltd., Suffolk, UK) or manually when confounding factors such as precipitation affected the accuracy of the automated counter. The background lawn was inspected for signs of toxicity. Data were confirmed to meet the following acceptability criteria: 1) the mean vehicle control counts fell within the laboratory’s historical 99% confidence intervals for group means and/or 2) each vehicle control plate count fell within the historical 99% reference ranges, and 3) the positive control plate counts were comparable with the historical 99% reference ranges.
2.3. in vitro Micronucleus assay
The methodology used in this study was based on draft OECD Test Guideline 487 (OECD, 2008). An appropriate volume of whole blood from two healthy, non-smoking male volunteers was drawn from the peripheral circulation into heparinized tubes within two days of culture initiation. Blood was stored refrigerated and pooled using equal volumes from each donor prior to use. Whole blood cultures were established in sterile disposable centrifuge tubes by placing 0.4 mL of pooled heparinized blood into 9 mL of HEPES-buffered RPMI medium containing 10% (v/v) heat inactivated fetal calf serum and 50 μg/mL gentamycin. Phytohaemagglutinin (PHA, reagent grade, Gibco, Loughborough, UK) was included in the culture medium at a final concentration of approximately 2% to stimulate the lymphocytes to divide. Replicate cultures (four for vehicle controls, two for positive controls and each test article concentration) were established for each test condition. Cultures were incubated at 37°C for 48 hours and rocked continuously. Immediately prior to treatment, 0.1 mL of culture medium was removed from the 24 hour continuous cultures to achieve a final pre-treatment volume of 9.3 mL. The S9 mix used for metabolic activation was prepared by mixing glucose-6-phosphate (180 mg/mL), NADP (25 mg/mL), KCl (150 mM) and rat liver S9 in the ratio 1:1:1:2. The final concentration of the liver homogenate in the test system was 2%; an equivalent volume of 150 mM KCl was added to cultures treated in the absence of S9. S9 mix or KCl (0.5 mL per culture) was added and the cultures were treated with perillaldehyde or controls (0.1 mL per culture). Cyclophosphamide (CPA; 12.5 μg/mL), mitomycin C (MMC; 0.08 μg/mL), and vinblastine (VIN; 0.02 μg/mL) were used as indirect and direct-acting positive controls. Cytochalasin B in DMSO (0.1 mL) was added to the 24 hour continuous cultures at the time of treatment. Cultures were incubated at 37°C for 3 (±S9) or 24 (-S9) hours. Following exposure, the 3 hour (±S9) cultures were centrifuged at 300 g for 10 minutes, washed twice with sterile saline (pre-warmed to 37°C), and resuspended in fresh pre- warmed medium containing fetal calf serum, gentamycin and Cytochalasin B at 6 μg/mL. These cultures were allowed to recover at 37°C for 21 hours. At the defined sampling time, cultures were centrifuged at 300 g for 10 minutes. Pelleted cells were resuspended in 4 mL of 0.075 M KCl at 37°C for 4 minutes to allow cell swelling to occur. Cells were then fixed by dropping the KCl suspension into fresh, cold methanol/glacial acetic acid (3:1, v/v). The fixative was changed by centrifugation (300 g, 10 minutes) and resuspension and the process repeated as necessary (1250 g, 2-3 minutes) until the cell pellets were clean.
Lymphocytes were maintained refrigerated in fixative until at least the following day to ensure adequate fixation. Cells were centrifuged and resuspended in a minimal amount of fresh fixative as required to produce a milky suspension. Several drops of cell suspension were gently spread onto multiple clean microscope slides and allowed to dry. The slides were stained for 5 minutes in filtered 4% (v/v) Giemsa in Gurr’s phosphate buffer, pH 6.8, rinsed, dried, and mounted with coverslips. Slides were examined for proportions of mono-, bi-, and multinucleate cells (500 cells/culture). Slides from the concentration at which approximately 55% (typically 50%-60%) reduction in replication index (RI; [number binucleate cells + 2(number multinucleate cells)]/total number of cells in treated cultures) had occurred and at least two lower concentrations were selected for microscopic analysis, such that a range of cytotoxicity from maximum (not excessive) to little was covered. Coded slides were analyzed by scoring 1000 binucleate cells from each culture (2000 per concentration) for micronuclei.
2.4. in vitro HPRT mutation assay in L5178Y mouse lymphoma cells
The methodology and fluctuation protocol used in this study complies with OECD Test Guideline 476 (OECD, 1997b). The stock of L5178Y tk+/- (3.7.2C) mouse lymphoma cells were originally provided by Dr. Donald Clive (Burroughs Wellcome Co.). Cells were cultured in RPMI medium containing 100 U/mL penicillin and streptomycin, 2.5 μg/mL amphotericin B, 0.5 mg/mL Pluronic, and 5 or 10% heat inactivated horse serum. For 3 hour exposures, at least 107 cells in a volume of 18.8 mL of RPMI medium (5% serum) were seeded in sterile 50 mL centrifuge tubes. For 24 hour exposures, at least 4 x 106 cells in a volume of 19.8 mL RPMI medium (10% serum) were seeded in 75 cm2 tissue culture flasks. For all exposure conditions, 0.2 mL of vehicle, perillaldehyde, or positive control solutions was added to the culture. The S9 mix used for metabolic activation was prepared by mixing glucose-6-phosphate (180 mg/mL), NADP (25 mg/mL), KCl (150 mM) and Aroclor 1254-induced rat liver S9 (Moltox, Boone, NC) in the ratio 1:1:1:2. The final concentration of the liver homogenate in the test system was 2%; an equivalent volume of 150 mM KCl was added to cultures treated in the absence of S9. Duplicate (single for positive controls) exposures (±S9) were performed in a final volume of 20 mL. After incubation for 3 hours at 37oC with gentle agitation or stationary incubation at 37°C for 24 hours, cultures were centrifuged at 200 g for 5 minutes, washed, and resuspended in 20 mL RPMI medium (10% serum). Cell densities were adjusted to 2 x 105 cells/mL and cells were transferred to flasks for growth throughout the expression period or were diluted and plated for survival assessment. For survival measurements, 0.2 mL of cells diluted to 8 cells/mL was placed into each well of two 96-well microtiter plates. The plates were incubated at 37oC in a humidified incubator with 5% v/v CO2 in air for 7 days until scoreable. Wells containing viable clones were visually identified using background illumination and counted. For mutation frequency assessment, cultures were maintained in flasks for 7 days to allow for expression of Hprt- mutations. Sub-culturing was performed as necessary to avoid exceeding 1 x 106 cells/mL, retaining at least 6 x 106 cells/flask if possible.
Cultures to be plated for viability and 6-thioguanine resistance were selected on the basis of observations of recovery and growth of the cultures during the expression period. Following the 7 day expression period, cell densities in the selected cultures were adjusted to 1 x 105 cells/mL. For viability assessment, samples from these were further diluted to 8 cells/mL, plated, and scored after 7 to 8 days as described above. For mutation frequency assessment, 6-thioguanine was added to the cultures to give a final concentration of 15 μg/mL and 0.2 mL of each suspension was transferred to each well of four 96-well microtiter plates. Plates were incubated until scoreable (12 to 14 days), and wells containing clones were identified and counted.
From the zero term of the Poisson distribution, the probable number of clones/well (P) on microtiter plates is given by the formula: P = -ln(empty wells/total wells). Plating efficiency (PE) of each culture was calculated as PE = P/number of cells plated per well. Percentage relative survival (% RS) in each test culture was determined by comparing plating efficiencies in test and control cultures: % RS = [PE(test)/PE(control)] x 100. Any loss of cells during the 3 hour exposure period was accounted for by adjusting the % RS values for each test article concentration as follows: adjusted % RS = % RS x (post-treatment cell concentration for test article treatment/post-treatment cell concentration for vehicle control). Mutant frequency (MF) is usually expressed as “mutants/106 viable cells”: MF = [PE(mutant)/PE(viable)] x 106. Since 2 x 104 cells were plated/well for mutation to 6-thioguanine resistance and an average of 1.6 cells were plated/well on viability plates, MF = [P(mutant)/2 x 104]/[P(viable)/1.6] x 106 = (-ln[empty wells/total wells(mutant)]/-ln[empty wells/total wells(viable)]) x 80.
2.5. in vivo Micronucleus/comet assay
2.5.1 Animal husbandry
Male Han Wistar rats (Charles River (UK) Ltd., Margate, UK) were 8-10 weeks of age at the time of treatment. Animals were housed in wire topped, solid bottomed cages (three animals per cage) with European softwood bedding (Datesand Ltd., Manchester, UK), in a temperature and humidity controlled AAALAC accredited facility with a 12 hour light/12 hour dark cycle. SQC Rat and Mouse Maintenance Diet No 1, Expanded (Special Diets Services Ltd., Witham, UK) and water were provided ad libitum. Animals were provided wooden Aspen chew blocks and rodent retreats for environmental enrichment and were acclimatized for at least 5 days prior to initiation of dosing.
2.5.2. Experimental design
A combined micronucleus and comet assay was conducted in accordance with OECD test guideline 474 (OECD, 1997c) for the micronucleus assay and current recommendations for the comet assay at the time the study was conducted (Burlinson et al., 2007; EFSA, 2012; Tice et al., 2000). A dose range-finding study was conducted to estimate the maximum tolerated dose (MTD) of perillaldehyde in male and female rats. Since no substantial gender differences in toxicity were revealed in the range-finder experiment, male rats were selected for the MN/comet assay. Five groups of male rats (6 rats/group) were administered perillaldehyde at dose levels of 175, 350, and 700 mg/kg/day (estimated to be 25% MTD, 50% MTD, and MTD, respectively), vehicle (corn oil), or the positive control compound, ethyl methanesulfonate in purified water at 150 mg/kg/day, by oral gavage daily for three days (0, 24 and 45 hours). Three hours after the final dose the animals were euthanized and femurs were collected for analysis of micronuclei in the bone marrow, liver and duodenum tissues were collected for the comet assay and histopathology, and blood samples were collected for clinical chemistry as described below.
2.5.3. Erythrocyte micronucleus assay
Femurs were cleaned of adherent tissue and the ends removed from the shanks. Using a syringe and needle, bone marrow was flushed from each femur with 2 mL of fetal bovine serum (FBS). The samples were filtered through cellulose columns containing an equal mix of type 50 and α-cellulose at 50 mg/mL. Once the majority of the FBS had passed through the column an additional 4 mL of FBS were added to the sample tubes and loaded onto the columns. The filtered bone marrow cells were centrifuged at 200 g for 5 minutes at 15-25°C and the cell pellets were gently resuspended in 3 mL of FBS and pelleted again; the supernatant was aspirated to leave one or two drops used to resuspend the cell pellet using a Pasteur pipette. One drop of cell suspension was placed on the end of each slide and smeared by drawing the end of a clean slide along the sample slide. Slides were air-dried prior to fixing for 10 minutes in absolute methanol, rinsed several times in distilled water, and air-dried. One slide per animal was immediately stained for 5 minutes in 12.5 μg/mL acridine orange in 0.1 M phosphate buffer pH 7.4. Stained slides were rinsed in phosphate buffer, then dried and stored protected from light at room temperature. At least 500 erythrocytes were analyzed for the relative proportions of polychromatic erythrocytes (PCE), visible as bright orange enucleated cells, and normochromatic erythrocytes (NCE), visible as smaller dark green enucleated cells. At least 2000 PCE/animal were examined for the presence of micronuclei. All slides were coded to eliminate scorer bias.
2.5.4. Comet assay
Liver samples were washed thoroughly in Merchants solution (0.5 mM NaEDTA, 10% DMSO in phosphate buffered saline, pH 7.4), cut into small pieces in fresh Merchants solution, and pushed through bolting cloth (pore size of 150 μm) with approximately 4 mL of Merchants solution to produce single cell suspensions. Duodenum samples were washed thoroughly in Merchants solution and vortexed in fresh Merchants solution for approximately 15 seconds. The tissue was removed from the Merchants solution and the inner surface gently scraped (released material discarded) using the back of a scalpel blade. The tissue was vortexed in Merchants solution for a further 15 seconds prior to gently scraping the inside of the duodenum with the back of a scalpel blade. All cell suspensions were maintained on ice prior to slide preparation. Four slides were prepared per tissue for each animal. Slides were dipped in molten normal melting point agarose, the underside of the slides was wiped clean, and the slides were allowed to dry. Each single cell suspension (40 μL) was added to 400 μL of 0.7% low melting point agarose at approximately 37°C and 100 μL of cell suspension/agarose mix was placed on each slide. The slides were then coverslipped and placed on ice. Once gelled, coverslips were removed and the slides placed in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10 [adjusted with NaOH], 1% Triton X-100, 10% DMSO) at 2-8°C protected from light. One slide for each animal was incubated in lysis buffer for 1 hour (“halo”); remaining slides (comet assay) were lysed overnight. Following lysis, the slides for the comet assay were washed in purified water for 5 minutes and transferred to electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH>13) at 2-8°C to allow the DNA to unwind for 20 minutes (duodenum) or 30 minutes (liver). The slides were electrophoresed in the same buffer at 0.7 V/cm for 20 minutes (duodenum) or 40 minutes (liver). A block design was employed for the unwinding and electrophoretic steps in order to avoid excessive variation across the groups for each electrophoretic run (i.e., the same number of slides was processed at a time for each animal). Following electrophoresis, slides were neutralized in 0.4 M Tris, pH 7.0 (3 x 5 minute washes), dried, and stored at room temperature. Following lysis, the “halo” slides were placed in 0.4 M Tris, pH 7.0 for approximately 3 x 5 minutes and then dried and stored as described above.
Immediately prior to scoring, coded slides were stained with 100 μL of 2 μg/mL ethidium bromide and coverslipped. Scoring was carried out using fluorescence microscopy at an appropriate magnification and with suitable filters. Comet scoring was performed using “Comet Assay IV” image analysis system (Perceptive Instruments, Suffolk, U.K.). Measurements of tail intensity (% DNA in tail) and Olive tail moment were generally obtained from at least 150 cells/animal scored on 2-3 slides. The number of “hedgehog” cells [a morphology indicative of highly damaged cells which can be associated with severe cytotoxicity, necrosis or apoptosis (Burlinson et al., 2007; Lorenzo et al., 2013; Tice et al., 2000); also known as “clouds” and “ghosts”] observed during comet scoring was tabulated for each animal but these cells were not scored as comets. Halo slides, used to evaluate cytotoxicity, were scored by assessing 100 cells/slide and recording the number with a clear halo of DNA around the nucleus. Acceptable cytotoxicity criteria were considered to be ≤30% hedgehogs and/or ≤30% cells with halos in the vehicle control.
2.5.5. Clinical chemistry and histopathology
Blood obtained from the abdominal aorta under deep anesthesia was collected into lithium heparin anticoagulant, mixed for two minutes, cooled, centrifuged at 2300 g for 10 min at 4°C, and plasma was analyzed for clinical chemistry parameters (see Table 7 for list of parameters). Samples of liver were preserved in neutral buffered formalin, embedded in paraffin wax, sectioned at 5 μm, and stained with haematoxylin and eosin. Slides were examined by the study pathologist and digitized whole slide scans underwent peer-review evaluation.
3.0. Statistical analyses
For the bacterial mutagenicity study, Dunnett’s test was used to compare the counts at each concentration with the vehicle control and the presence of a concentration-dependent response was assessed. The test was considered positive for mutagenicity if a reproducible concentration related significant response (p ≤ 0.01) was induced. For the in vitro micronucleus test, the proportions of binucleate cells with micronuclei in the replicate samples were used to establish acceptable heterogeneity between replicates by means of a binomial dispersion test. The proportion of binucleate cells with micronuclei for each treatment condition was compared with the proportion in negative controls by using Fisher’s exact test. Probability values of p ≤ 0.05 were considered significant. The criteria for a positive response in the assay were a statistically significant increase in the frequency of binucleate cells with micronuclei at one or more concentrations that exceeded the normal range in both replicates, and observation of a concentration-related increase in the proportion of binucleate cells with micronuclei. For the in vitro HPRT mutation assay, the log mutant frequency for each treatment concentration was compared with that of the control cells and the data were evaluated for a concentration-dependent response in mutant frequency correlating with perillaldehyde treatment. For these tests, the heterogeneity factor was calculated to obtain a modified estimate of variance. The test article was considered to induce forward mutation at the Hprt locus in L5178Y lymphoma cells if the mutant frequency at one or more concentrations was significantly greater than that of the negative control (p≤0.05), there was a significant linear trend analysis (p≤0.05), and the effects were reproducible.
For the in vivo micronucleus assay, inter-individual variation in the numbers of MN-PCE was estimated for each group by means of a heterogeneity chi-square calculation. The numbers of MN-PCE in each treated group were compared with the numbers in vehicle control groups using a 2×2 contingency table to determine chi-square. The tests were interpreted with one-sided risk for increased frequency with increasing dose. Probability values of p ≤ 0.05 were considered significant. A linear trend test was used to evaluate possible dose-response relationships. For the comet assay, Levene’s test for equality of variances across the groups was performed (p ≤ 0.01). Heterogeneous data were rank-transformed prior to analysis. The data were analyzed separately using one-way analysis of variance (ANOVA). An overall dose response test was performed along with Dunnett’s test for pairwise comparisons of each treated group with the vehicle control. All tests were performed with one-sided risk for increased response with increasing dose. The positive control group was compared to the vehicle control using a two sample t-test. The test was interpreted with one-sided risk for increased response with increasing dose. Criteria for a positive result in the micronucleus and comet assays were at least one statistically significant dose group, a positive dose group falling outside the range of laboratory historical control data, and a statistically significant dose response trend. A test was considered equivocal if only one or two of these conditions were met.
3. Results
3.1. Results of the bacterial reverse mutation assay
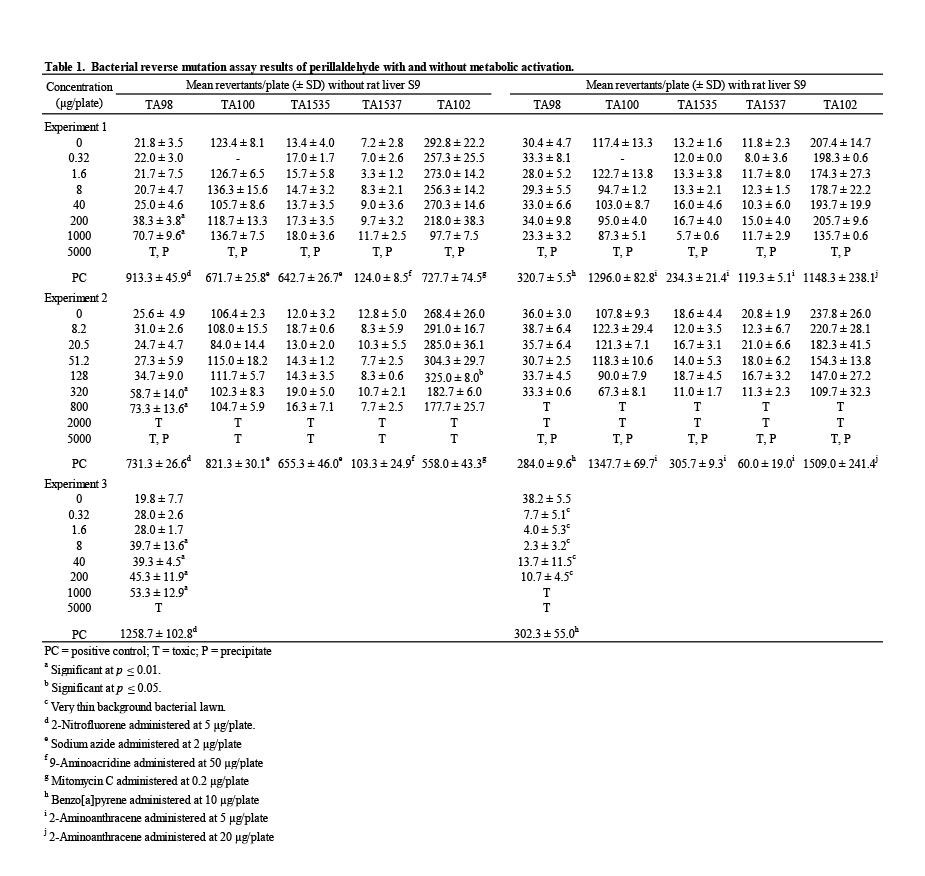
Mutagenicity assays were conducted to assess the potential of perillaldehyde to induce gene mutations in bacteria (Table 1). A range-finding assay using TA100 provided evidence of toxicity and precipitation at 5000 μg/plate; therefore, a top concentration of 5000 μg/plate, with and without metabolic activation, was chosen as recommended by OECD (OECD, 1997a). The initial mutagenicity test used concentrations of perillaldehyde ranging from 0.32 to 5000 μg/plate in a plate incorporation assay. Following these treatments, evidence of toxicity (ranging from a slight thinning of the background bacterial lawn and/or reduction in revertant numbers to a complete killing of bacteria) was observed at 1000 and/or 5000 μg/plate in all strains in the absence and presence of S9 (±S9). Precipitation was observed at 5000 μg/plate in all strains ±S9. A statistically significant and concentration-related increase in revertant numbers was observed in strain TA98 at 200 and 1000 μg/plate in the absence of S9 (Table 1, Experiment 1). No mutagenic effect of perillaldehyde was detected in the other strains ±S9.
In a second mutagenicity test, narrowed concentration intervals were employed covering the range 8.2 – 5000 μg/plate to better examine concentrations of perillaldehyde approaching the maximum test concentration; in addition, all treatments in the presence of S9 included a pre- incubation step to increase the sensitivity for detection of mutagenic chemicals in this assay system. In this experiment, evidence of toxicity was observed at 320, 800, and/or 2000 μg/plate and above in all strains in the absence and/or presence of S9. Precipitation was observed at 5000 μg/plate in all strains +S9 and in TA98 –S9. This experiment produced results similar to those of the first experiment—a statistically significant and concentration-related increase in revertant numbers in strain TA98 in the absence of S9 was observed with no evidence of mutagenic activity in the other strains ±S9 (Table 1, Experiment 2).
To confirm the reproducibility of the increased revertants observed in the first two experiments, a third mutagenicity assay was performed in TA98 ±S9, using a different batch of perillaldehyde and the same treatment conditions and concentrations employed in the first experiment. Exposure to perillaldehyde at 5000 μg/plate in the absence of S9 resulted in complete killing of the bacteria; in the presence of S9 extensive toxicity was evident by very thin background bacterial lawns and marked reductions in the numbers of revertants at all concentrations up to 200 μg/plate, and complete killing of the bacteria at the 1000 and 5000 μg/plate concentrations. No precipitation was observed on the test plates. Following exposure of strain TA98 to perillaldehyde in the absence of S9, statistically significant increases in revertant numbers were measured at concentrations ≥ 8 μg/plate. Therefore, the induction of revertants in strain TA98 was reproduced and is considered to be evidence of in vitro mutagenic activity in this strain. No statistically significant increases in revertant numbers were observed for any other strain tested. The vehicle and positive controls for all experiments met acceptance criteria.
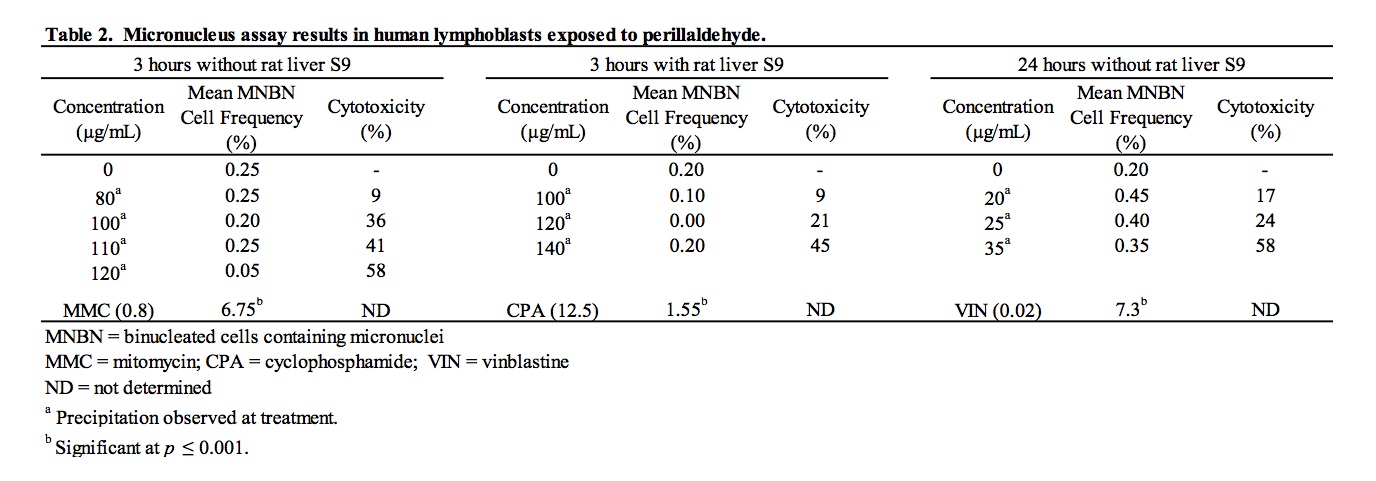
3.2. Results of the in vitro micronucleus assay
A suitable range of concentrations of perillaldehyde to be tested for micronucleus induction was selected on the basis of results of a preliminary range finder toxicity experiment. Micronucleus frequency and cell viability data for cultures exposed to perillaldehyde are summarized in Table 2. Steep concentration-related toxicity was observed between 140 and 160 μg/mL, resulting in 45% and 71% cytotoxicity, respectively. Consequently, 140 μg/mL was close to a concentration producing a 50%-60% reduction in RI and was considered to be the maximum concentration suitable for analysis. Treatment of cells with perillaldehyde in the absence and presence of S9 under all treatment conditions resulted in frequencies of micronucleated binucleate cells that were similar to and not significantly different from those measured in concurrent vehicle controls for all concentrations analyzed. The micronucleated binucleate cell frequency of perillaldehyde-treated cultures fell within, or slightly below, laboratory historical ranges. Exposure to cyclophosphamide, used as a positive control chemical requiring metabolic activation, and mitomycin C and vinblastine, included as direct-acting positive controls, led to significant (p ≤ 0.001) induction of MN as compared to the vehicle controls. It is concluded that perillaldehyde did not induce micronuclei in cultured human peripheral blood lymphocytes when tested at (or very close to) the limit of cytotoxicity in both the absence and presence of S9.
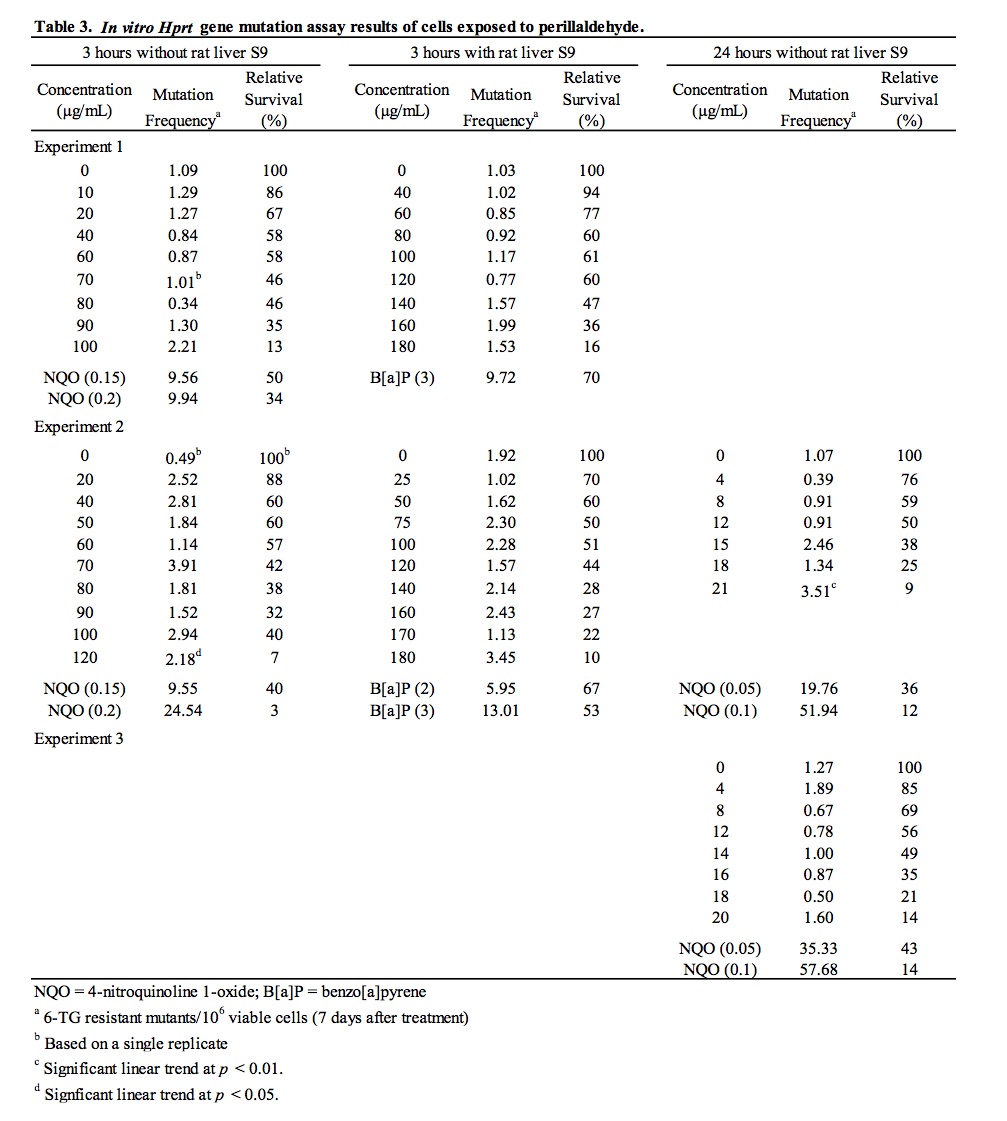
3.3. Results of the in vitro mouse lymphoma mutation assay
Following cytotoxicity range-finder experiments to determine concentrations of perillaldehyde providing >10% relative survival under different test conditions, perillaldehyde was assayed for the ability to induce mutation at the hypoxanthine-guanine phosphoribosyl transferase (Hprt) locus [6-thioguanine resistance] in mouse L5178Y lymphoma cells. Results of the HPRT mutation assay are summarized in Table 3. An initial experiment was conducted for 3 hours in the absence and presence of metabolic activation. The highest concentrations analyzed to determine viability and 6-thioguanine resistance were 100 μg/mL in the absence of S9 and 180 μg/mL in the presence of S9, which resulted in 13% and 16% relative survival, respectively. A second experiment was conducted for 3 and 24 hours in the absence of S9 and 3 hours in the presence of S9. None of the tested concentrations for the 3 and 24 hour (-S9) conditions produced 10-20% relative survival; therefore, the highest concentration selected to determine viability and 6-thioguanine resistance for the 3 and 24 hour (-S9) conditions were 120 μg/mL (which resulted in 7% relative survival) and 21 μg/mL (which resulted in 9% relative survival), respectively. The highest concentration analyzed for the 3 hour (+S9) condition was 180 μg/mL, which resulted in 10% relative survival. Subsequently, a third experiment was conducted for 24 hours in the absence of S9 to clarify the equivocal results for this treatment condition obtained in the previous experiment. In the third experiment, the highest concentration analyzed to determine viability and 6-thioguanine resistance was 20 μg/mL, which resulted in 14% relative survival. In all three experiments, no statistically significant increases in mutant frequency were detected following exposure to perillaldehyde at any concentration tested in the absence and presence of metabolic activation. Statistically significant linear trends were measured following 3 and 24 hour exposures in the absence of S9 in the second experiment; however, there were no statistically significant increases in mutant frequency at any concentration evaluated under these conditions. The small, sporadic increases in the magnitude of mutant frequency and the positive linear trend observed in the second experiment were not reproduced in the third experiment. In all three experiments, mutant frequencies in vehicle control cultures fell within acceptable ranges based on laboratory historical data and significant increases in mutation were induced by the positive control chemicals, 4-nitroquinoline 1-oxide (-S9) and benzo(a)pyrene (+S9). Since none of the concentrations tested in three independent experiments induced mutations, and the positive trends measured for 3 and 24 hour exposures in the absence of S9 were not reproduced over two experiments, it is concluded that perillaldehyde did not induce mutation at the Hprt locus of L5178Y mouse lymphoma cells under the conditions tested in this study.
3.4. Results of the in vivo MN/comet assays
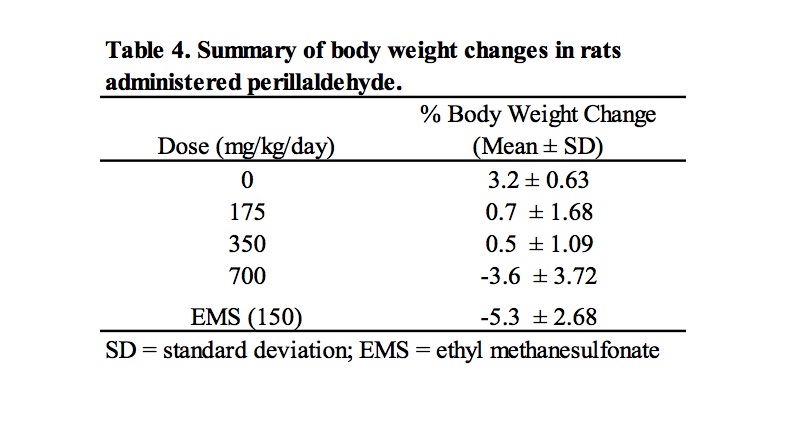
Based on the results of a preliminary dose setting study, a combined MN/comet assay was conducted in which male Han Wistar rats were administered perillaldehyde at 175, 350, and 700 mg/kg/day for 3 consecutive days. Clinical signs of toxicity were limited to animals dosed at 700 mg/kg/day, for which reduced levels of activity were observed in 5/6 animals; in addition, one animal displayed symptoms of ataxia and a second animal exhibited piloerection. Dose related decreases in body weight gain or weight loss were observed at all dose levels (Table 4).
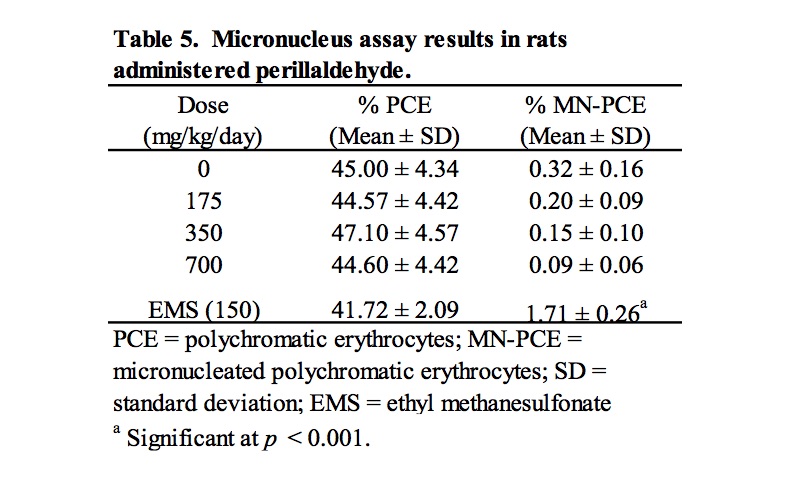
Results of analysis of MN-PCE and PCE frequencies are summarized in Table 5. Rats treated with perillaldehyde at all doses exhibited group mean %PCE that were similar to the concurrent vehicle control group, indicating a lack of bone marrow toxicity. There were no statistically significant increases in MN-PCE for any of the groups treated with perillaldehyde compared to the concurrent vehicle control, the micronucleus frequencies were considered consistent with the laboratory’s historical vehicle control data, and the positive control chemical, ethyl methanesulfonate, elicited a statistically significant response. Thus, under the conditions of this study, perillaldehyde did not induce an increase in MN-PCE of the bone marrow of male rats following oral gavage administration of doses up to 700 mg/kg/day.
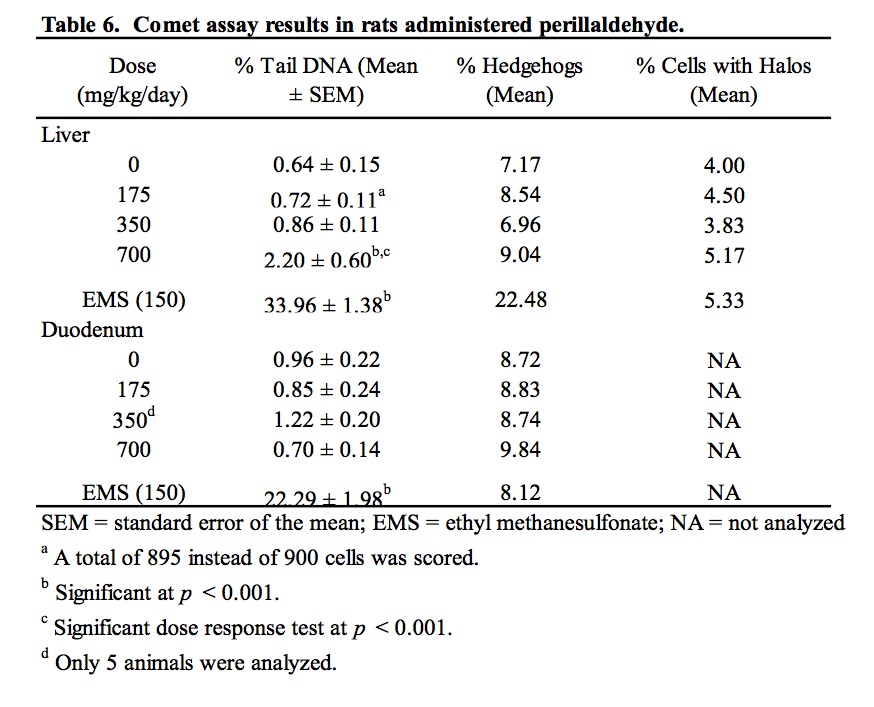
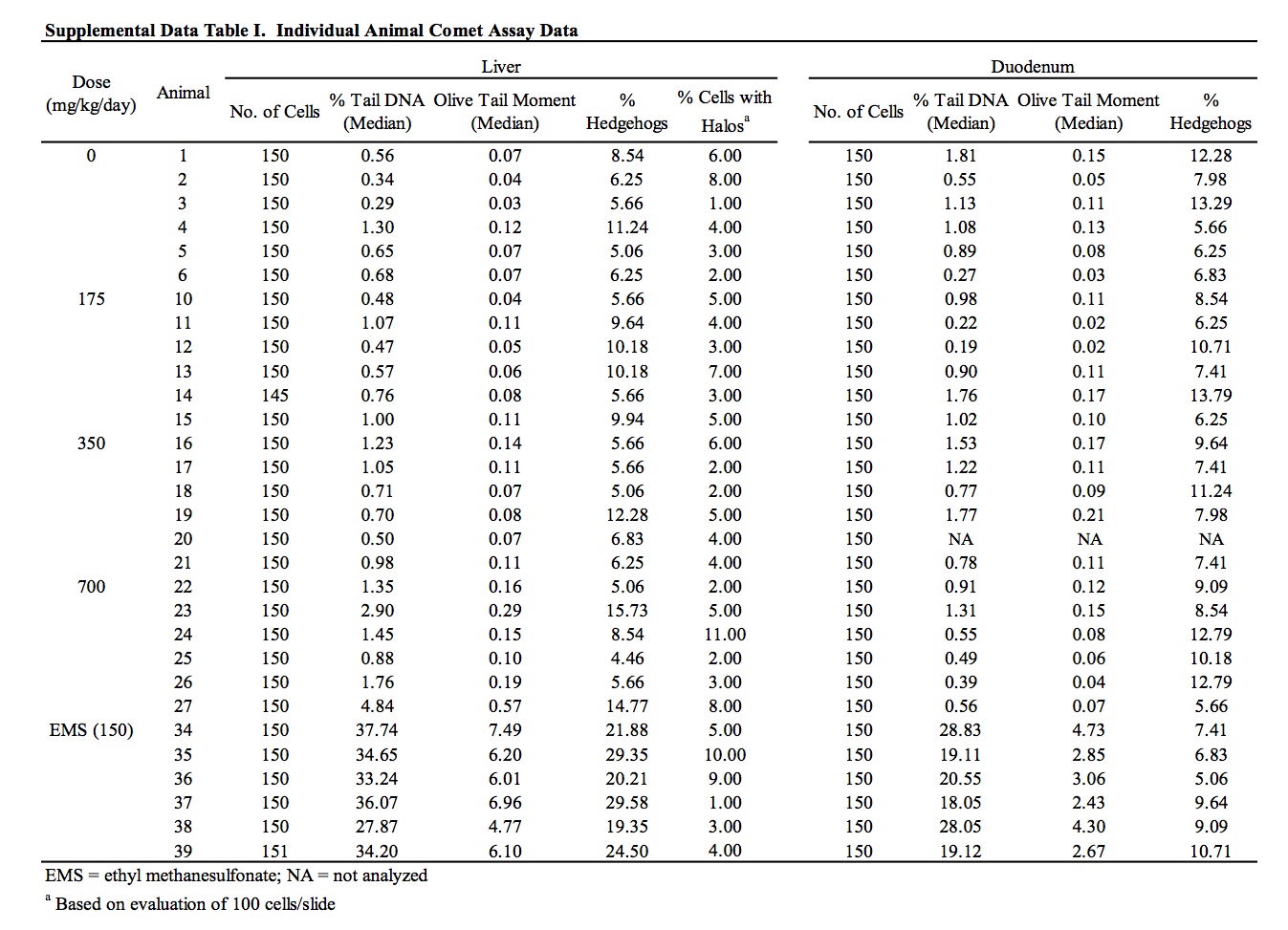
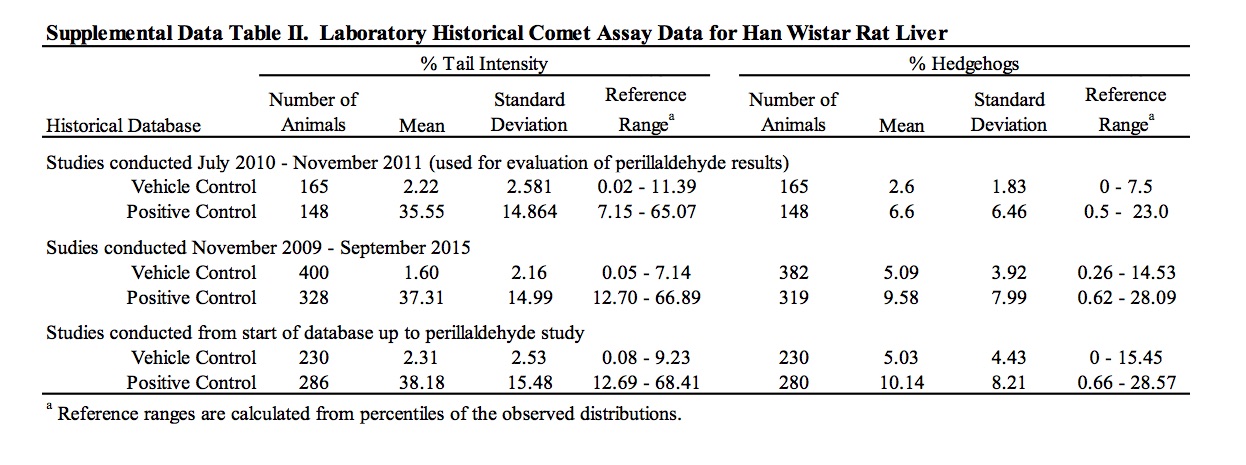
The results of the assessment of DNA damage in two potential target organs of rats administered perillaldehyde, as measured by the comet assay, are provided in Table 6 and Supplemental Data Table I. There was no evidence of DNA damage induced by perillaldehyde in the duodenum of exposed rats. Group mean % tail intensity values for the liver of animals in the 175 and 350 mg/kg/day dose groups were not significantly different from the concurrent vehicle control. At the highest dose (700 mg/kg/day), a small but statistically significant increase in liver % tail intensity was observed. A statistically significant linear trend was also measured. Damage due to chemical-related cytotoxicity or to excessive mechanical disruption during cell isolation can confound interpretation of comet assay results. In this study, there was no dose-related increase in % hedgehogs or % cells with halos in liver cells of exposed animals. Five out of the six animals in the 700 mg/kg/day dose group had % tail intensity values that exceeded those of the concurrent vehicle control animals. However, all six animals fell well within the laboratory’s historical vehicle control 95th percentile range (0.02 – 11.39; n = 165) and only two of the six animals had % tail intensity values that exceeded the mean % tail intensity of the historical control data (2.22%). Administration of the positive control chemical, ethyl methanesulfonate, induced a statistically significant increase in DNA damage in both liver and duodenum falling well outside of the confidence range of historical vehicle control data.
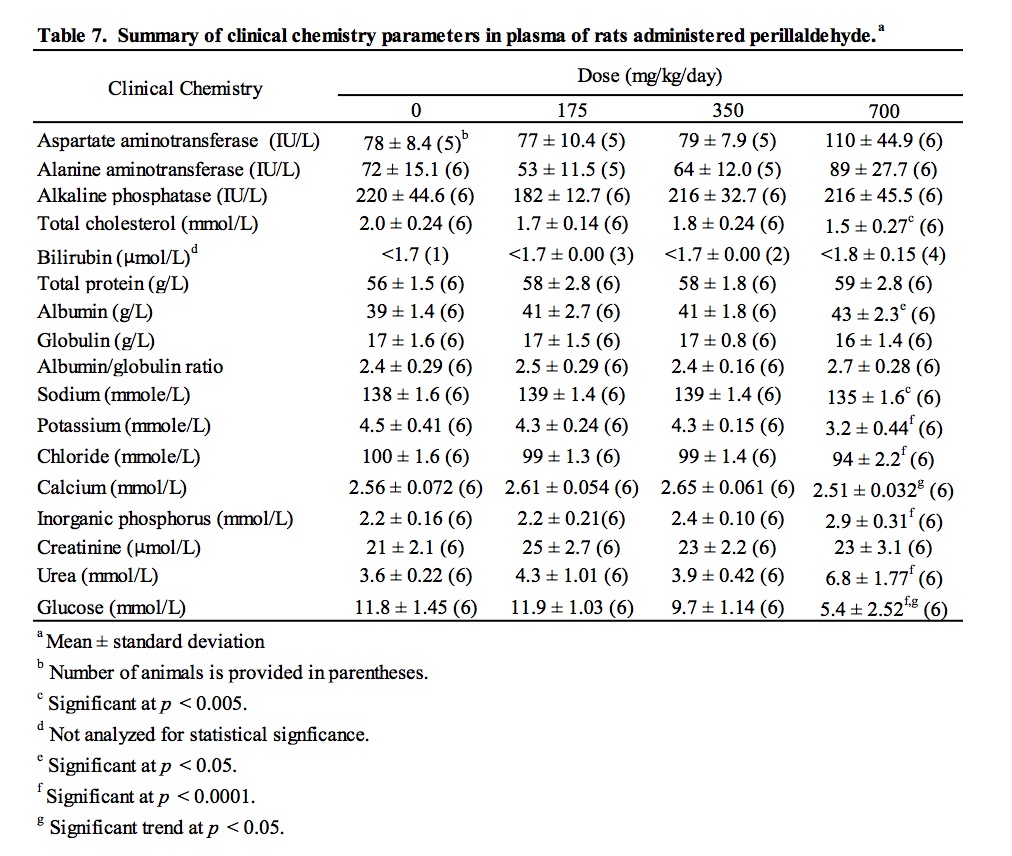
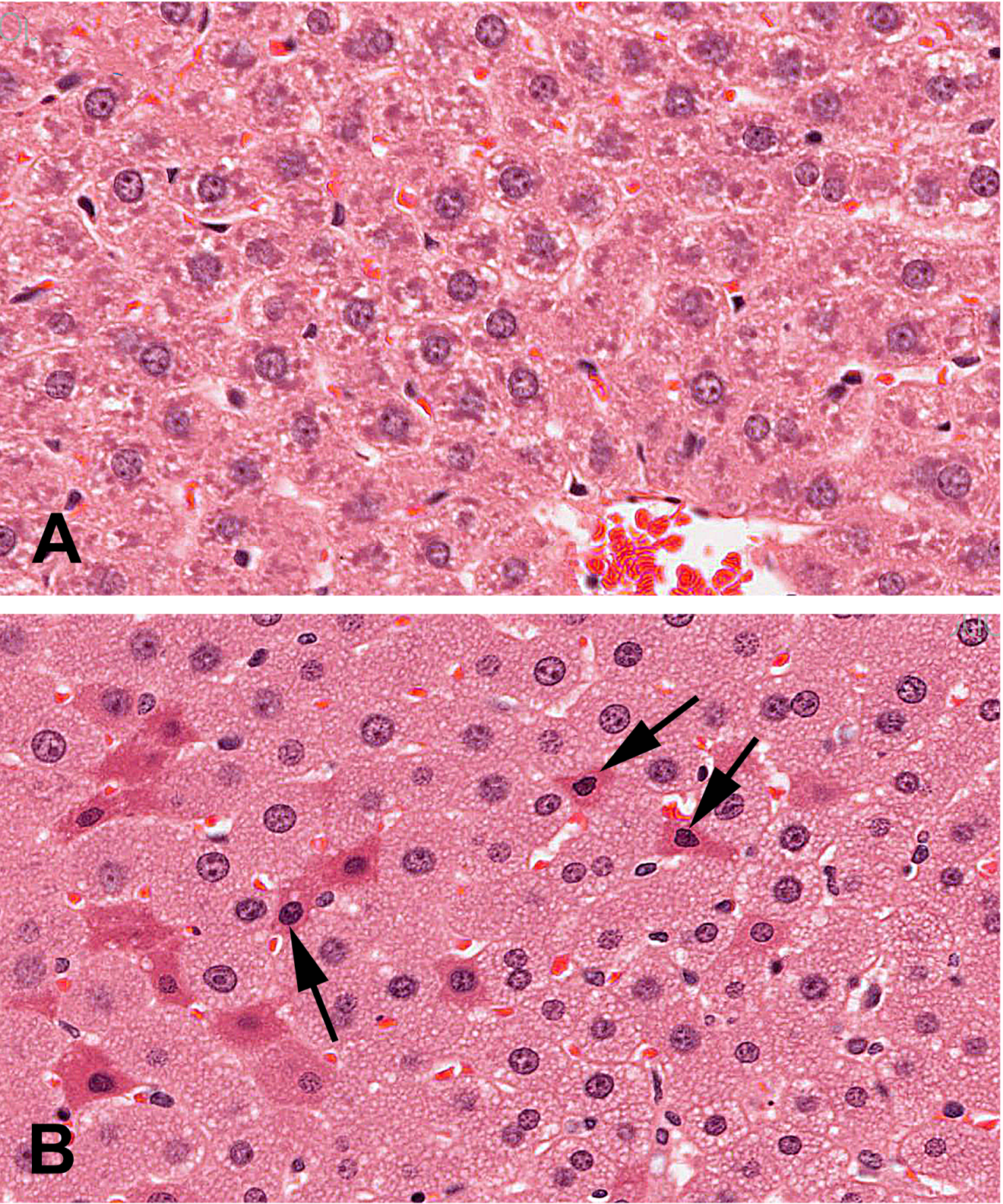
There were no macroscopic findings considered to be related to administration of perillaldehyde. On microscopic examination, hepatocytes in the liver of animals administered perillaldehyde at 700 mg/kg/day were enlarged due to the presence of multiple small cytoplasmic vacuoles consistent with microvesicular fat deposits with attendant compression of sinusoidal channels (Fig. 2). This vacuolation was predominantly periportal to mid-lobular in localization, but sometimes affected the centrilobular hepatocytes and was present in all six rats in the perillaldehyde dose group. Centrilobular hepatocyte degeneration characterized by loss of cytoplasmic and nuclear detail was occasionally evident in rats in the 700 mg/kg/day dose group, typically in areas in which cytoplasmic vacuolation was less severe. Individual as well as small clusters of shrunken hepatocytes with condensed nuclear chromatin and hypereosinophilic cytoplasm were present to varying degrees only in this dose group. During clinical chemistry assessment of blood samples it was noted that a high number of samples were lipemic; this may be attributed to the corn oil used as the vehicle for test article formulation, which was administered just three hours prior to blood sampling. As a consequence, some samples were deemed unsuitable for statistical analysis of certain parameters. Measurements were obtained for at least five animals/group for aspartate aminotransferase (AST) and alanine aminotransferase (ALT); although mean serum values for these enzymes were not statistically significant, three of the six rats dosed with 700 mg/kg/day perillaldehyde exhibited high activity of both ALT and AST, indicative of hepatic toxicity. Statistically significant findings supportive of general toxicity that were present only in the rats in the 700 mg/kg/day dose group included increased serum urea and decreases in serum cholesterol, glucose, sodium, chloride, and potassium (Table 7).
4. Discussion
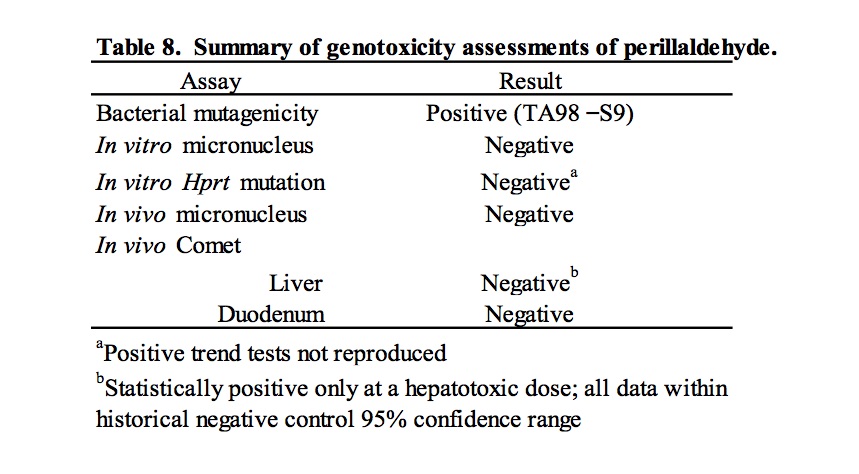
These studies were conducted in response to a request by EFSA for genotoxicity data for flavouring substances considered representative of groups of compounds containing α,β- unsaturated aldehyde and ketone structures, or their potential precursors. Specifically, the genotoxicity of perillaldehyde was evaluated as a representative substance of FGE.19 subgroup 2.2. The ability of perillaldehyde to induce mutations, chromosomal abnormalities, and DNA damage was evaluated in accordance with the test strategy outlined by EFSA for evaluation of FGE.19 subgroup substances (EFSA, 2008a). The results of these tests are summarized in Table 8. In a bacterial reverse mutation assay, perillaldehyde induced mutation in histidine-requiring Salmonella typhimurium strain TA98 in the absence of S9. This result differs from the previously published negative results for a study conducted in a variety of bacterial strains (Ishidate et al., 1984). However, it is unclear if the treatment conditions for these studies are comparable. Moreover, the published results were reported as “– or +”, so observation of a weak response (<2x the concurrent control) cannot be ruled out. In an in vitro micronucleus assay testing perillaldehyde in human lymphocytes, no induction of micronuclei formation was detected in a short (3 hour) exposure in the presence and absence of metabolic activation or in a long (24 hour) exposure in the absence of metabolic activation. These results do not support published positive results of a chromosome aberration study (Ishidate et al., 1984) that should be interpreted with caution due to lack of detail regarding cytotoxicity.
The negative result in the rat bone marrow micronucleus assay reported here is consistent with a previous report in which perillaldehyde was administered to male ddY mice by i.p. injection in single or four daily doses (Hayashi et al., 1988). The duodenum was evaluated in the comet assay as a first site of contact in rats exposed to perillaldehyde by oral gavage; there was no evidence of induced DNA damage. Administration of 700 mg/kg/day perillaldehyde did cause a small, statistically significant increase in DNA damage in the liver. and a statistically positive trend. The international effort to validate the in vivo comet assay for the detection of genotoxic carcinogens, coordinated by the Japanese Center for the Validation of Alternative Methods (JaCVAM), concluded that histopathology remains the “gold standard” for assessing tissue cytotoxicity, and changes in % tail DNA require careful interpretation when measured in conjunction with severe histopathological changes (Uno et al., 2015). Hepatocyte cytoplasmic vacuolation was observed in liver sections from the rats exposed to 700 mg/kg/day perillaldehyde. The small vacuoles are morphologically consistent with microvesicular fat. Shrunken hepatocytes with condensed nuclear chromatin and hypereosinophilic cytoplasm, typically associated with early stages (i.e., apoptosis/necrosis) in the cell death pathway, were also observed in the top dose group. The shrunken hepatocytes are probably secondary to anoxia from compression of hepatic sinusoids by the enlarged vacuolated hepatocytes. Small increases in liver enzymes (AST and ALT) in the 700 mg/kg/day dose group, accompanying the hepatocellular vacuolation, are consistent with hepatotoxicity. Thus, the very small increase in % tail intensity observed following exposure to 700 mg/kg/day perillaldehyde may have been caused by hepatotoxicity rather than direct damage resulting from interaction of perillaldehyde with DNA. Indeed, a false positive finding in the comet assay resulting as an indirect consequence of liver toxicity has been demonstrated previously. Chloroform, a non-genotoxic carcinogen and a well-known hepatotoxicant, tested positive in the liver with associated histopathology changes, including hepatocellular vacuolation and enlarged hepatocytes, in the JaCVAM validation trial (Uno et al., 2015). As such, to ensure consistency with OECD guidelines, conclusions regarding the ability of perillaldehyde to damage DNA should be based on concentrations that do not result in overt liver toxicity that confound interpretation of the data.
The use of laboratory historical control data is important for considering the biological significance of a positive result in a genotoxicity assay. The OECD guideline for the in vivo comet assay states that criteria for a positive test should include a result lying outside the distribution of matched historical negative control data (OECD, 2014). In the comet assay of perillaldehyde, the % tail intensity for all of the animals in the positive dose group fell at the low end of the 95% reference range of laboratory historical negative control data calculated on the basis of a robust (n = 165) dataset. Furthermore, the group mean % tail intensity for the statistically significant perillaldehyde dose group was equivalent to the mean value in the historical control database. Notably, the OECD test guideline for the comet assay also indicates that, on the basis of the JaCVAM validation, the group mean vehicle control values in the liver preferably should not exceed 6%. The perillaldehyde comet findings (both the group mean and individual animal data) are well within this upper limit for acceptable vehicle control values, further supporting the conclusion that the small increase in tail intensity observed in the liver following administration at 700 mg/kg/day was not biologically relevant and was most likely an artefact of the observed hepatic cytotoxicity. Notably, if the positive test in the liver, the major site of metabolism in vivo, is assumed to reflect a biologically relevant result, then it is surprising that in vitro mutagenicity and chromosome damage tests performed in the presence of metabolic activation did not provide any indication of genotoxic effects. Placed in the context of potential human risk, the daily exposure for a person in Japan weighing 55.1 kg is approximated to be 0.0011 mg/kg body weight/day, calculated on the basis of estimated perillaldehyde consumption data for FY2011 (Ueda and Sato, 2015); based on these estimates the margin of safety for human exposure to perillaldehyde relative to the 700 mg/kg body weight/day that tested statistically positive in rats in the comet assay is approximately five orders of magnitude.
The results of these genotoxicity studies have been reviewed by EFSA which determined perillaldehyde to be negative in the in vitro micronucleus test and positive in TA98 in the Ames test without a metabolic activation system (EFSA, 2013). However, contrary to the conclusions stated here and within the study reports, EFSA determined the results of the in vitro HPRT mutation assay and in vivo comet assay as equivocal and positive, respectively (EFSA, 2013, 2015). EFSA determined the outcome of the HPRT mutation assay to be equivocal on the basis of positive linear trends measured in one experiment that were not reproduced in the other experiments. EFSA’s criticism of using the historical control data for interpretation of the significant result in the comet assay is based on the fact that the vehicle control 95% reference range in use at the time of the study was sufficiently wide to overlap the positive control reference range, and therefore, may not be appropriate for use as an evaluation criterion. In that regard, it is useful to consider the perillaldehyde positive % tail intensity value in the context of broadened historical datasets collated by the testing laboratory, in which the vehicle and positive control reference ranges are clearly distinct (Supplemental Data Table II). Comparison of the positive % tail intensity value (2.20%) to historical data that included the studies performed immediately prior to the perillaldehyde study (n = 230) or historical data spanning the period immediately prior and subsequent to the perillaldehyde study (n = 400) confirms that the perillaldehyde data are close to the means of both vehicle control data sets (2.31% and 1.60% respectively), supporting the arguments that the positive comet result for perillaldehyde is not biologically relevant.
Based on concern about genotoxicity in the liver, EFSA concluded that perillaldehyde demonstrated genotoxic potential in vivo and was a potential safety concern as a flavouring substance (EFSA, 2015). Following this evaluation, the European Commission published notification of its intent to remove this flavouring substance from the EU list of flavourings (http://eur-lex.europa.eu/eli/reg/2015/1760/oj). Previously, JECFA evaluated perillaldehyde as a member of a group consisting of cyclic primary alcohols, aldehydes, acids, and related esters, and concluded that its low level consumption as a flavouring agent poses no safety concern (JECFA, 2003). Because substances belonging to this group are expected to be quickly metabolized into less toxic substances, JECFA concluded that they are not of genotoxic concern in vivo, and JECFA has not currently reevaluated their risk based on the new available data. Perillaldehyde was classified as a GRAS substance by FEMA in 1978 (Oser and Ford, 1978). Following the recent EFSA assessment, the FEMA Expert Panel reviewed the results of the comet study reported here. It concluded that the results did not rationalize a change in GRAS status because changes were observed only at the maximum dose which was associated with signs of general toxicity and hepatotoxicity, and because the changes fell within the range of laboratory historical data (Cohen et al.; publication in preparation).
In summary, although there is some in vitro indication of genotoxic potential in the absence of metabolic activation, consensus opinion is that the micronucleus and comet assays in rats do not provide credible evidence of in vivo genotoxic potential resulting from exposure to perillaldehyde, supporting its safe use in food and beverages. These results reinforce the original conclusions of JECFA and the more recent reevaluation by the FEMA Expert Panel that perillaldehyde poses no safety concern for humans when used as a flavouring agent and consumed at estimated dietary exposures. Furthermore, by extrapolation on the basis of structural similarities (EFSA, 2008b), it seems unlikely that the other α,β-unsaturated alicyclic aldehyde flavourings categorized as FGE.19 subgroup 2.2 would pose a genotoxic risk to humans.
Acknowledgements
These studies were conducted at Covance Laboratories Ltd. and funded by the International Organization of the Flavor Industry (IOFI). ILS, Inc. was contracted to write the manuscript. Maronpot Consulting LLC, a consultant for the Japan Flavor and Fragrance Materials Association, provided pathology peer-review assessment of liver histopathology.
Figure Legend
Figure 1. Chemical structure of perillaldehyde. Perillaldehyde is a naturally occurring monocyclic terpenoid with an α,β-unsaturated aldehyde structure.
Figure 2. Photomicrographs of liver from vehicle control rat and rat exposed to the highest dose (700 mg/kg/day) of perillaldehyde. (A) Vehicle control: clear areas in cytoplasm of normal hepatocytes represent glycogen. (B) High dose rat: hepatocytes are enlarged with collapse of sinusoidal spaces and cytoplasmic vacuolation consistent with microvesicular fatty change. Individual degenerating hepatocytes are shrunken with hyper-eosinophilic cytoplasm and condensed nuclear chromatin (denoted with arrows).
References
Burlinson, B., Tice, R.R., Speit, G., Agurell, E., Brendler-Schwaab, S.Y., Collins, A.R., Escobar, P., Honma, M., Kumaravel, T.S., Nakajima, M., Sasaki, Y.F., Thybaud, V., Uno, Y., Vasquez, M., Hartmann, A., 2007. Fourth International Workgroup on Genotoxicity testing: results of the in vivo Comet assay workgroup. Mutat Res 627, 31-35.
EFSA, 2008a. Statement of the Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF) on Genotoxicity Test Strategy for Substances belonging to Subgroups of FGE.19. EFSA Journal 854, 1-5.
EFSA, 2008b. Statement of the Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF) on List of alpha, beta-Unsaturated Aldehydes and Ketones representative of FGE.19 substances for Genotoxicity Testing. EFSA Journal 910, 1-7.
EFSA, 2012. Minimum Criteria for the acceptance of in vivo alkaline Comet Assay Reports. EFSA Journal 10, 2977.
EFSA, 2013. EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids Scientific Opinion on Flavouring Group Evaluation 208 (FGE.208): Consideration of genotoxicity data on representatives for 10 alicyclic aldehydes with the ,- unsaturation in ring/side-chain and precursors from chemical subgroup 2.2 of FGE.19. EFSA Journal 11, 3151.
EFSA, 2015. EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids Scientific Opinion on Flavouring Group Evaluation 208 (FGE.208Rev1): Consideration of genotoxicity data on representatives for 10 alicyclic aldehydes with the ,-unsaturation in ring/side-chain and precursors from chemical subgroup 2.2 of FGE.19. EFSA Journal 13, 4173.
Hayashi, M., Kishi, M., Sofuni, T., Ishidate, M., Jr., 1988. Micronucleus tests in mice on 39 food additives and eight miscellaneous chemicals. Food Chem Toxicol 26, 487-500.
Ishidate, M., Jr., Sofuni, T., Yoshikawa, K., Hayashi, M., Nohmi, T., Sawada, M., Matsuoka, A., 1984. Primary mutagenicity screening of food additives currently used in Japan. Food Chem Toxicol 22, 623-636.
JECFA, 2003. Safety evaluation of certain food additives. Fifty-ninth meeting of the Joint FAO/WHO Expert Committee on Food Additives. WHO Food Additive Series, no. 50. http://www.inchem.org/documents/jecfa/jecmono/v50je01.htm.
Ji, W.W., Wang, S.Y., Ma, Z.Q., Li, R.P., Li, S.S., Xue, J.S., Li, W., Niu, X.X., Yan, L., Zhang, X., Fu, Q., Qu, R., Ma, S.P., 2014. Effects of perillaldehyde on alternations in serum cytokines and depressive-like behavior in mice after lipopolysaccharide administration. Pharmacol Biochem Behav 116, 1-8.
Lorenzo, Y., Costa, S., Collins, A.R., Azqueta, A., 2013. The comet assay, DNA damage, DNA repair and cytotoxicity: hedgehogs are not always dead. Mutagenesis 28, 427-432.
OECD, 1997a. OECD Guideline for the Testing of Chemicals: Bacterial Reverse Mutation Test. 471.
OECD, 1997b. OECD Guideline for the Testing of Chemicals: In Vitro Mammalian Cell Gene Mutation Test. 476.
OECD, 1997c. OECD Guideline for the Testing of Chemicals: Mammalian Erythrocyte Micronucleus Test. 474.
OECD, 2008. OECD Guideline for the Testing of Chemicals: In Vitro Mammalian Cell Micronucleus Test. 487.
OECD, 2014. OECD Guideline for the Testing of Chemicals: In Vivo Mammalian Alkaline Comet Assay. 489.
Omari-Siaw, E., Zhu, Y., Wang, H., Peng, W., Firempong, C.K., Wang, Y.W., Cao, X., Deng, W., Yu, J., Xu, X., 2016. Hypolipidemic potential of perillaldehyde-loaded self- nanoemulsifying delivery system in high-fat diet induced hyperlipidemic mice: Formulation, in vitro and in vivo evaluation. Eur J Pharm Sci 85, 112-122.
Oser, B.L., Ford, R.A., 1978. Recent progress in the consideration of flavoring ingredients under the Food Additives Amendment.11. GRAS substances. Food Technology 32, 60-70.
Tian, J., Wang, Y., Zeng, H., Li, Z., Zhang, P., Tessema, A., Peng, X., 2015. Efficacy and possible mechanisms of perillaldehyde in control of Aspergillus niger causing grape decay. Int J Food Microbiol 202, 27-34.
Tice, R.R., Agurell, E., Anderson, D., Burlinson, B., Hartmann, A., Kobayashi, H., Miyamae, Y., Rojas, E., Ryu, J.C., Sasaki, Y.F., 2000. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35, 206-221.
Ueda, H., Sato, H., 2015. Research on daily exposure estimate based on poundage data of food additives. JAFAN 35, 1-38.
Uno, Y., Kojima, H., Omori, T., Corvi, R., Honma, M., Schechtman, L.M., Tice, R.R., Beevers, C., De Boeck, M., Burlinson, B., Hobbs, C.A., Kitamoto, S., Kraynak, A.R., McNamee, J., Nakagawa, Y., Pant, K., Plappert-Helbig, U., Priestley, C., Takasawa, H., Wada, K., Wirnitzer, U., Asano, N., Escobar, P.A., Lovell, D., Morita, T., Nakajima, M., Ohno, Y., Hayashi, M., 2015. JaCVAM-organized international validation study of the in vivo rodent alkaline comet assay for detection of genotoxic carcinogens: II. Summary of definitive validation study results. Mutat Res Genet Toxicol Environ Mutagen 786-788, 45-76.
Xu, L., Li, Y., Fu, Q., Ma, S., 2014. Perillaldehyde attenuates cerebral ischemia-reperfusion injury-triggered overexpression of inflammatory cytokines via modulating Akt/JNK pathway in the rat brain cortex. Biochem Biophys Res Commun 454, 65-70.