Microarray gene expression analysis offers great promise to help us understand the molecular events of experimental carcinogenesis, but have such promises been fulfilled? Studies of gene expression profiles of rodent are being published and demonstrate that yes, indeed, gene array data is furthering our understanding of tumor biology. Recent studies have identified differentially expressed genes in rodent mammary, colon, lung, and liver tumors. Although relatively few genes on the rodent arrays have been fully characterized, information has been generated to better identify signatures of histologic type and grade, understand invasion and metastasis, identify candidate biomarkers of early development, identify gene networks in carcinogenesis, understand responses to therapy, and decifer overlap with molecular events in human cancers. Data from mouse lung, mammary gland, and liver tumor studies are reviewed as examples of how to approach and interpret gene array data. Methods of gene array data analysis were also applied for discovery of genes involved in the regression of mouse liver tumors induced by chlordane, a nongenotoxic murine hepatocarcinogen. Promises are beginning to be fulfilled and it is clear that pathologists and toxicologists, in collaboration with molecular biologists, bioinformatists, and other scientists are making great strides in the design, analysis, and interpretation of microarray data for cancer studies.
INTRODUCTION
There is an explosion of studies utilizing microarray gene expression analysis to explore the molecular pathways involved in toxicity and disease, and many of these studies are focused on finding the alterations involved in carcinogenesis. There are numerous studies examining gene pro- files in human cancer and relatively few utilizing rodent neoplasms. Understanding the genetic pathways of cancer in rodent models offers promise to prevent and successfully treat cancer in all species. Several recent studies in the rodent have identified differentially expressed genes in mammary (Desai et al., 2002a, 2002b; Simpson et al. 2003; Green et al. 2004), colon (Chen et al., 2003), lung (Lin et al., 2001; Yao et al., 2002; Bonner et al., 2004) and liver tumors (Masui et al., 1997; Graveel et al., 2001; Meyer et al., 2003; Liu et al., 2004). In these studies investigators have identified gene expression signatures of histologic type and grade, pathways involved in invasion and metastasis, candidate biomarkers of early development, potential oncogenes and tumor suppressor genes, gene networks in carcinogenesis, responses to therapy, and overlap with molecular events in human cancers. Examples of such findings in the mouse as well as data from a mouse liver tumor regression study will be presented as examples of how gene array studies are furthering our understanding of the carcinogenic process.
MOLECULAR PROFILING OF MOUSE NEOPLASIA
Lung Tumors
Studies of chemically induced mouse lung carcinogenesis in the laboratory of Ming You (Bonner et al., 2003, 2004) have identified distinct genetic differences between normal lung and mouse lung neoplasms, as well as between mouse lung adenomas and carcinomas. Using a mouse array containing sequences that represent about 12,000 genes, they identified an expression profile of 20 genes that distinguished lung adenomas and carcinomas from normal lung (Bonner et al., 2004). Some of the altered genes identified are known cancer genes (i.e., the putative tumor suppressor gene APC2 (van Es et al., 1999) showed relatively decreased expression in the neoplasms). Fifty genes were found that had exactly the opposite profiles of expression when comparing lung adenomas to carcinomas; thus, identifying genes and pathways with potential importance in the progression of benign lung neoplasia to malignancy (Bonner et al., 2004). The expression profiles of lung tumors can potentially be used as an adjunct to diagnose, classify, and grade lung neoplasms.
By comparing the gene profiles in the mouse lung neoplasms to the 1345 genes differentially expressed in embryonic and fetal lung development, 25 genes that are similarly or oppositely dysregulated in lung cancer were identified (Bonner et al., 2004). Since many oncogenes are overexpressed and tumor suppressor genes repressed in cancer cells, and such genes are similarly changed in embryological and fetal development, it can be deduced that some or all of these 25 newly found genes are either oncogenes (n = 3) or tumor suppressor genes (n = 22). This finding also substantiates the theory that the development of cancer can recapitulate the development of an organ. After identifying the homologous gene sequences between the genes on the mouse array with those in a human lung cancer array study, a hierarchical clustering analysis1 revealed 39 altered genes with similar profiles in mouse and human lung cancer (Bonner et al., 2004). Many of these 39 genes are known or suspected to play a role in the carcinogenic process and others are good candidates for further investigation. For the hundreds of genes other than those 39, the gene expression varied greatly between the species and between individual human lung neoplasms. This suggests there are heterogenous responses as measured by gene array and multiple, as well as common, pathways in cancerous growth. By hierarchical clustering analysis1 the investigators also found evidence that mouse bronchoalveolar adenomas were more similar in genetic profile to human low grade lung adenocarcinomas and that mouse adenocarcinomas were more similar to the higher grade and more aggressive human large cell carcinomas and adenocarcinomas (Bonner et al., 2004). This type of information is valuable in the interpretation of animal models of cancer and furthering our understanding of the comparative patholobiology of cancer.
Mammary Tumors
Genetic profiles of mammary tumors of various genetically altered mice have revealed common genetic pathways as well as specific new insights on tumor heterogeneity in progression and “oncogenic signatures” (Kavanaugh and Green, 2003; Green et al., 2004). In the laboratory of Jeff Green at NCI, gene expression profiles of mammary neoplasms from 6 well-characterized transgenic mouse models of breast cancer (MMTV-c-myc, MMTV-neu, MMTV-Ha-ras, MMTVpolyoma middle T-ag (PyMT), C3(1)/SV40, and WAP-SV40/ T-ag) revealed that, despite different initiating events, the gene expression profiles were remarkably similar to each other, and they shared changes found in human breast cancer (Desai et al., 2002a, 2002b). Altered genes in the mice (that are also in human breast cancer) included those involved in carcinogenic processes such as cell cycle control, tumor cell adhesion, angiogenesis, apoptosis, and metabolism.
Small subsets of unique genes (“oncogenic signatures”) were differentially expressed in the MMTV-c-myc (3% of the genes were unique); MMTV-neu, MMTV-Ha-ras, and PyMT mice (7% of the genes were unique); and T-ag mice (19% of the genes were unique) (Desai et al., 2002b). For example cyclin B1 was unique to the mice overexpressing SV40 T-antigen (T-ag) suggesting this was a specific gene-pathway alteration and a potential therapeutic target for neoplasms driven by T-ag. T-ag is known to inactivate the tumor suppressor genes pRB and p53, which can be mutated in human breast cancer (Desai et al., 2002b). In another study using the neu/S100A4 transgenic model of metastatic breast cancer (Simpson et al., 2003), there was marked intertumoral heterogeneity of gene expression profiles when comparing multiple primary tumors and metastases among the animals. The finding suggests that, despite their similar histological appearance and initiating genetic event, there are multiple pathways of cancer progression. Some of the genetic alterations in this model are likely good candidate biomarkers of prognosis.
Liver Tumors
The liver transcriptome (total population of mRNAs) is second only to brain in its size and complexity (Shackel et al., 2002). It is believed that about 25–45% of all genes are expressed in this heterogeneous organ and that the transcriptome doubles or triples in complexity during disease states such as cancer (Shackel et al., 2002; Malarkey et al., 2005). There are a number of recent studies examining the gene profiles of hepatocellular tumors in mice. In a transplacental arsenic carcinogeneisis study in C3H male mice by Liu et al. (2004) differentially expressed gene expression profiles were found in hepatocellular neoplasms (82 genes representing ∼14% of the genes on the array) as well as nontumorous liver from treated mice (60 genes representing ∼10% of the genes on the array). The latter finding in nonneoplastic liver suggests that there are permanent gene alterations in adulthood as a result of in utero exposure to a carcinogen. Genes found altered in nontumorous liver from treated mice included c-myc, H-ras, α-fetoprotein, superoxide dismutase, glutathione-S-transferase (GST), Bcl-2, caspase 8, insulin-like growth factor binding protein 1 (IGFBP1), and estrogen receptor-α, many of which were also altered in the neoplasms (Table 1) (Liu et al., 2004). Together the findings highlight and/or confirm that dysregulation of the insulin-like growth factor (IGF) axis (Lee et al., 1997; Gong et al., 2000; Scharf et al., 2000; Scharf et al., 2001; Price et al., 2002; Yakar et al., 2002) and overexpression of cyclin D1 (Anna et al., 2003) are involved in the carcinogenic process. Studies are underway at the National Toxicology Program (NTP) to examine other hepatic carcinogens that might have a similar long-term effect on nonneoplastic liver.
Gene expression analysis of liver tumors from C3H mice treated once neonatally with diethylnitrosamine (DEN) by Graveel et al. (2001) revealed 194 genes with altered expression out of the ∼6500 on the microarray, some of which are known to be altered in human hepatocellular carcinoma. Genes dysregulated in the neoplasms included H19 and IGFBP1, CD63, intestinal trefoil factor 3 (Tff3), monokine induced by γ interferon (MIG), and osteopontin (Table 1). Novel genes, called cancer related gene-liver 1 (CRG-L1) (Graveel et al., 2001) and cancer related gene-liver 2 (CRGL2) (Graveel et al., 2003), were discovered and have since been proposed to be biomarkers of malignant liver neoplasia in mice and humans.
Meyer et al. (2003) examined gene expression in hepatocellular carcinomas from 26 peroxisomal fatty acyl-CoA oxidase null mice (AOX−/−) (Fan et al., 1996), 3 C57BL/6 J male mice treated with ciprofibrate (a synthetic peroxisome proliferator that acts through PPARα); and 3 C57BL/6 J mice treated with DEN. Of the approximately 500 genes detected as differentially expressed on the cDNA microarray (containing about 9000 gene sequences) among all groups of mice, there were 37 that were found in common among all 3 groups (Meyer et al., 2003). Included among the differentially expressed genes were some implicated in cancer such as lipocalin 2 (Bratt, 2000; Xu and Venge, 2000; Seth et al., 2002), IGFBP1 (Lee et al., 1997; Scharf et al., 2001), cathepsins (Navab et al., 1997 Bank et al., 2000; Koblinski et al., 2000; Krepela, 2001; Nasu et al., 2001), CRG-L1 (Graveel et al., 2001), and CD63 (Graveel et al., 2001) (Table 1). Hierarchical clustering revealed that the profiles of the tumors from the AOX −/− were very similar to the mice treated with ciprofibrate and different than those from mice treated with DEN, suggesting that the nongenotoxic PPARα cancer pathways differ from those involved with genotoxic pathways of DEN.
Genetics of Liver Tumor Regression
We have shown previously that lesions diagnosed as either hepatocellular adenomas and carcinomas in mice treated with chlordane regress after cessation of the chlordane administration (Malarkey et al., 1995). Upon cessation of chronic exposure to chlordane (55 ppm) in B6C3F1 mice at least 30% of hepatocellular adenomas and carcinomas regress (Malarkey et al., 1995). Chlordane was introduced in the 1940s as the first chlorinated cyclodiene insecticide and used extensively for the control of numerous agricultural pests until banned in the United States, Canada, and Western Europe in the 1980s. Chlordane is a nongenotoxic murine hepatocarcinogen that is believed to be associated with increased risk of reproductive problems, immune dysfunction, and cancer. It is listed as “possibly carcinogenic to humans” (IARC group 2B) and identified by the United Nations Environmental Program as 1 of the top 12 persistent organic pollutants requiring urgent attention (Malarkey et al., 1995; Yang and Chen, 1999; Huang and Chen, 2004).
Spontaneous tumor regression has been reported in a number of cases of histologically benign and malignant human neoplasms, including the liver (Lam et al., 1982; Challis and Stam, 1990; Gaffey et al., 1990; Chien et al., 1992; Papac, 1998; Takeda et al., 2000; Ikeda et al., 2001). For example, regression of hepatocellular carcinomas has not only been reported in mice after cessation of chlordane exposure, but also in rats after cessation of nafenopin (Grasl-Kraupp et al., 1997), in children after cessation of growth hormone supplementation that contained androgens (McCaughan et al., 1985; Fremond et al., 1987), and in women after ceasing oral contraceptives (Emerson et al., 1980; Steinbrecher et al., 1981). Hepatocellular adenomas and/or altered hepatocellular foci have regressed in rats after cessation of phenobarbital, clofibrate (Greaves et al., 1986), or the peroxisome proliferator, WY-14,643 (Marsman and Popp, 1994).
The mechanisms at play are uncertain, however chlordane, phenobarbital, nafenopin, clofibrate, and WY-14,643 are all considered nongenotoxic murine carcinogens and experimental evidence indicates that each likely contributes to the carcinogenic process, at least in part, by altering the balance of genes that regulate apoptosis (Grasl-Kraupp et al., 1997; Diez-Fernandez et al., 1998; Christensen et al., 1999). For example, nontumorous liver and/or the majority of liver neoplasms in mice treated with chlordane, phenobarbital, or WY 14,643 frequently express higher levels of inhibitory apoptotic proteins (such as Bcl-2 and Bcl-xL) (Christensen et al., 1999). It is probable that stopping the exposure to compounds like chlordane favors apoptotic pathways and regression of the neoplasms.
Gene microarray technology was applied in the study of gene expression and discovery of new genes involved in the regression of mouse liver tumors induced by chlordane. Chlordane was administered in the feed at 55 ppm to 210 B6C3F1 male mice and interim sacrifices were performed beginning at 408 and up to 570 days of age for mice treated continuously with chlordane or after cessation of treatment (63 mice) at 491 days (Malarkey et al., 1995). Analysis of gene expression was performed using NIEHS cDNA microarray mouse chips containing 8736 genes or expressed sequence tags (ESTs) as previously described (Iida et al., 2003). Briefly, total RNA was extracted from ∼0.4 gm each of 12 archived snap-frozen hepatocellular carcinomas, 6 from the continuously treated and 6 from the stop group. Three tumors of each group (stop and continuous) were either from mice at 22 or 77 days after the cessation of chronic exposure beginning at 491 days of age. mRNA
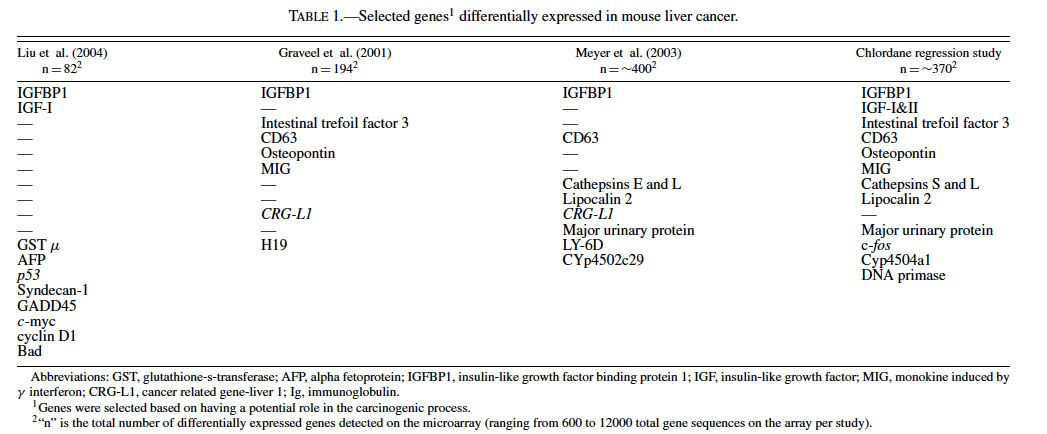
Using MAPS analysis (Bushel et al., 2001), a total of 370 genes of the approximately 9000 sequences on the array were differentially expressed and of those 7 appeared in 4 of the 6 comparisons, 30 in 3 of the 6 comparison, and the rest only appeared in 1 or 2 times out of the 6 paired samples (Table 1, Figure 1). Differential expression for 6 out of 6 genes chosen for validation was confirmed by nonisotopic RNase protection assay (c-fos, Cyp450 4A14, and cyr 61) or quantitative RT-PCR (osteopontin, cathepsin L, and an EST similar to goliath protein). Further validation was supported by concordant results for genes that had multiple sequences per gene on the microarray, such as Cyp450 4A14. Among the 370 genes many are proposed or known to be involved in carcinogenesis (cell growth, apoptosis, and progression), immune function, immediate early and stress responses, and lipid metabolism. For example, cathepsins L, S, and C were relatively higher in some of the neoplasms from the continuously exposed mice. Cathepsins, which are among the family of papain cysteine proteases, have mitogenic properties, inhibit apoptosis, and contribute to progression, invasion, and metastasis via matrix degradation properties (Navab et al., 1997; Bank et al., 2000; Koblinski et al., 2000; Krepela, 2001; Nasu et al., 2001). Osteopontin, which was expressed at greater levels in some tumors from the continuous groups, has been found overexpressed in rodent and human hepatocellular carcinomas (Graveel et al., 2001; Gotoh et al., 2002; Qin and Tang, 2004). Osteopontin has been called the “metastasis gene” involved in the spreading, migration, and adhesion of neoplastic cells (Pan et al., 2003; Tang et al., 2004). It promotes angiogenesis, inhibits apoptosis, and facilitates ras gene.
Comparing the list of differentially expressed genes found in mouse liver neoplasms (relative to normal, quiescent liver) from the Graveel et al. (2001) study (n = 194 genes) to the total gene list from the ∼9000 gene array used in chlordane regression study revealed an overlap of 16 genes (Figure 2), 11 of which were found when comparing the Graveel, et al list only to the 370 genes found my MAPS analysis in the regression study. Hierarchical clustering analysis revealed a profile that was dependent on the duration of time since cessation of chronic chlordane exposure after 491 days of age (Figure 2).
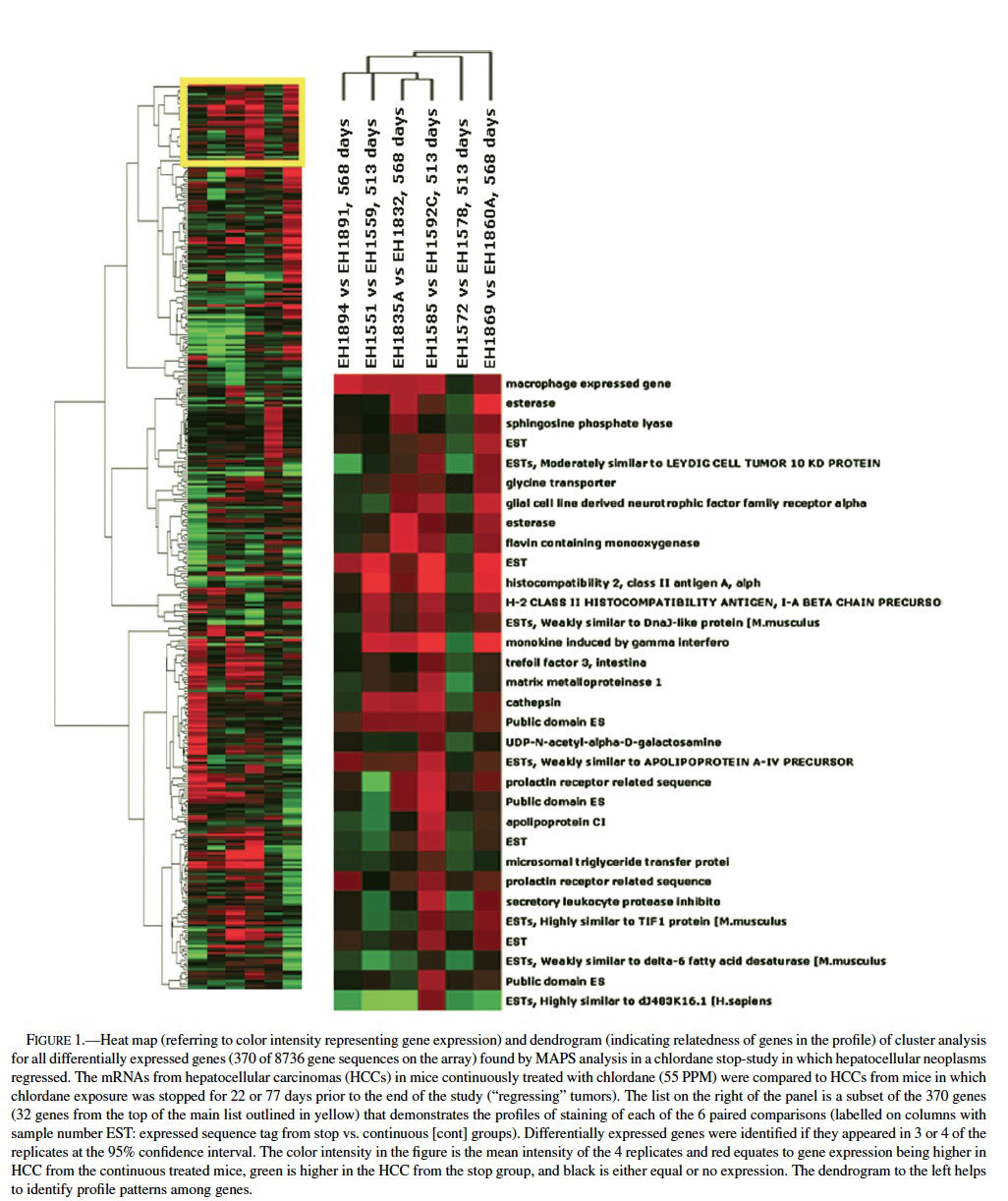
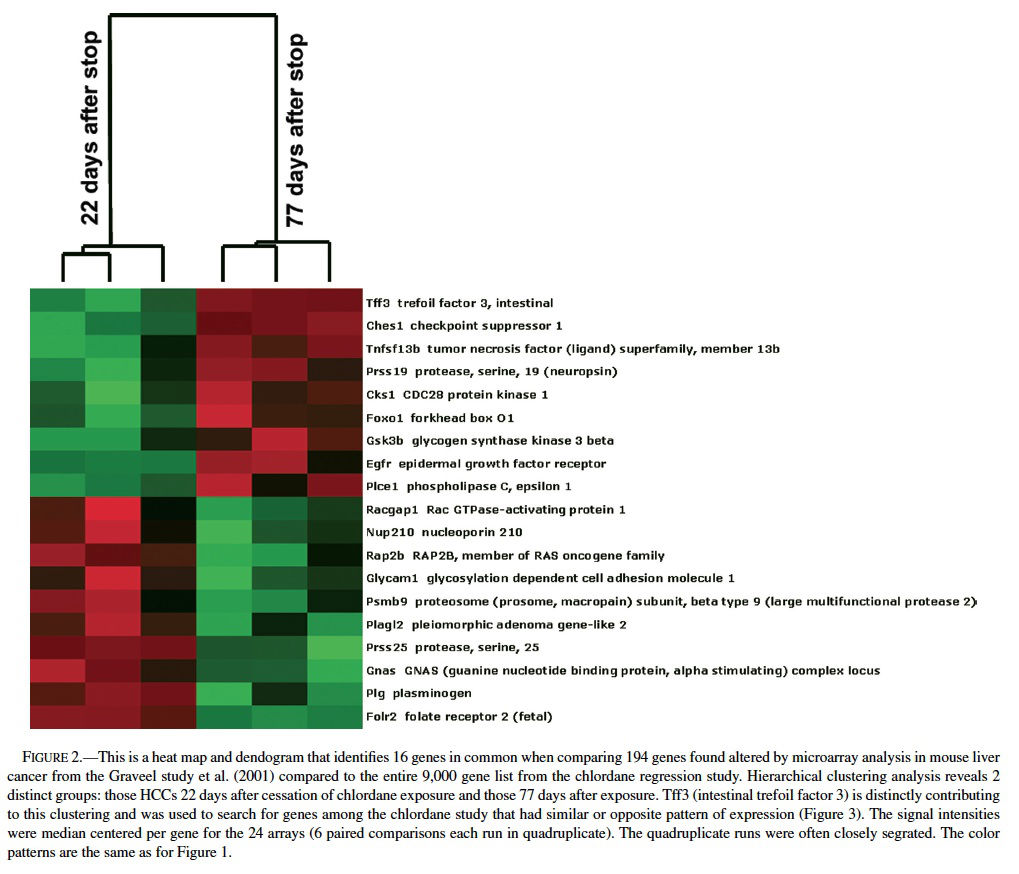
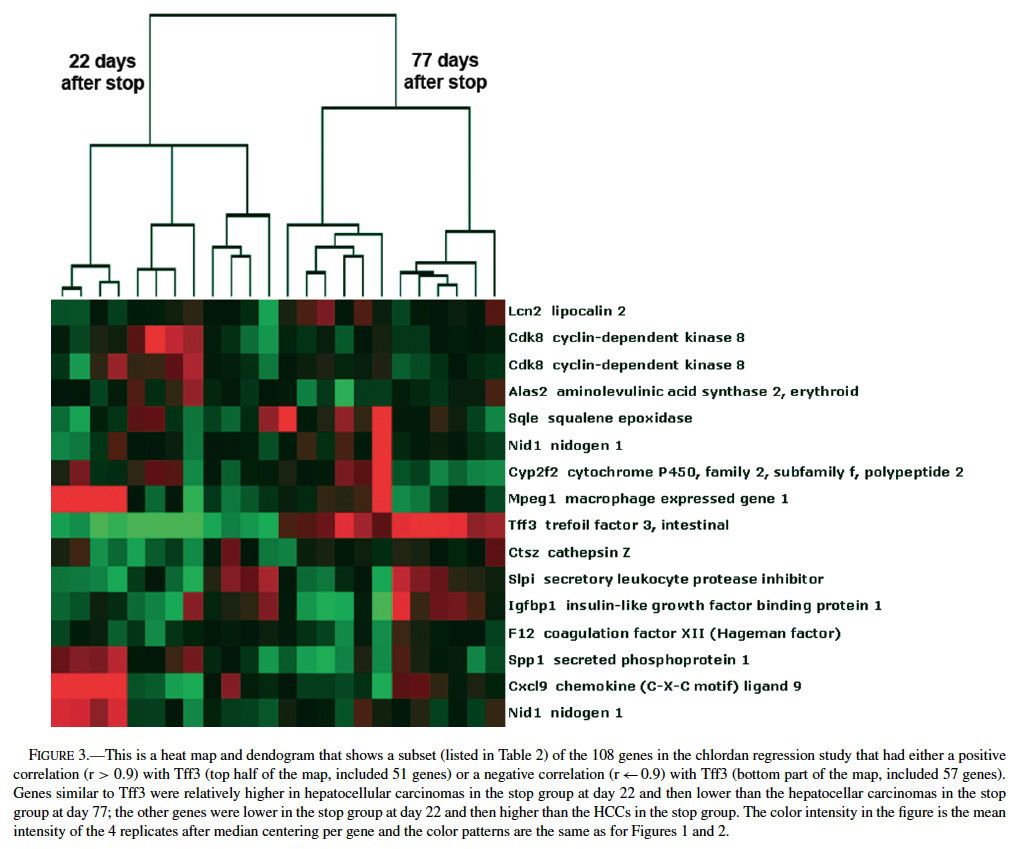
The gene profiles separated into two distinct groups, those 22 days or 77 days after cessation of chlordane exposure. The clustering appeared to be mainly driven by intestinal trefoil factor 3 (Tff3), which was relatively higher in hepatocellular carcinomas in the stop group at day 22 and then lower in the hepatocellular carcinomas in the stop group at day 77. Tff3 is a gastrointestinal peptide involved in mucosal injury and repair and acts as a motogen, stimulating epithelial migration (Emami et al., 2001; May and Westley, 1997a, 1997 b; Wright et al., 1997). It has been found to be overexpressed in ulcers, inflammatory bowel disease, and some gastrointestinal and breast cancers and has been proposed as a tumor suppressor in stomach cancer. Functionally, Tff3 inhibits apoptosis through an EGFR pathway and induces cell scattering and invasion of the basement membrane in cancer (May and Westley, 1997a, 1997b). The expression of Tff3 is regulated by FGF2, bFGF, estrogen, FGF7, and some other stimuli.
Because of the distinct expression profile for Tf3, we performed a supervised clustering analysis to search for genes with the same or opposite expression patterns as Tff3 (i.e., correlation values of r > 0.90 or r < −0.90) among the ∼9000 genes differentially expressed in the chlordane tumor regression study. We found 57 genes with similar and 51 with opposite patterns of Tff3 expression (Figure 3 and Table 2). Among these genes, some highlighted by EASE analysis, were some already known to be involved in carcinogenesis including glycogen synthase kinase 3 βgoGSK3β) (Desbois-Mouthon et al., 2002; Leis et al., 2002); TNF ligand superfamily member 13b (Schneider et al., 1999); Forkhead box 01 (Nakae et al., 2002); serine protease 25 (van Loo et al., 2002); plasminogen (Currier et al., 2003); pleomorphic adenoma gene-like 2 (Plagl2) (Furukawa et al., 2001). All of these are believed to have some role(s) in the promotion or prevention of cell death and some have been shown to act through the IGF pathways. GSK3β plays a role in the Wnt signaling pathway (Leis et al., 2002) that involves partnering with APC proteins and β-catenin; inhibits apoptosis; acts upstream of IGF and has been found to be dysregulated in HCC cell lines (Desbois-Mouthon et al., 2002). β-catenin mutations have been commonly demonstrated in mouse and human hepatocellular carcinomas (Devereux et al., 1999). Plagl2 is believed to be a tumor suppressor because it has been shown to have antiproliferative effects on tumors and promotes cell cycle arrest and apoptosis (Furukawa et al., 2001).

Summary of Mouse Liver Tumor Genetics
Genomics studies in liver carcinogenesis are helping us identify the many and complex genetic pathways involved in the development, progression, and even regression of neoplasia. Systematic comparison of data between laboratories leads to new clues and validation of findings and sheds light on important pathways and discovery of novel cancer genes. For example, repeated findings across mouse liver cancer studies have revealed potentially critical roles for c-fos, osteopontin, IGF, IGFBP1, Tff33, cathepsins (L,S, and C), CD63, monokine induced by γ interferon (MIG), as well as novel genes such as cancer related gene-liver 1 and as yet, uncharacterized genes. Along with the previously known genetic alterations shown in mouse liver, such as mutation in H-ras or β-catenin, overexpression of the Bcl-2 family of proteins (controlling apoptosis), TGFα, TGFβ, IGF II, EGFR, c-myc, raf, and cyclin D1 (Maronpot et al., 1995; Grisham, 1997; Chiaverotti et al., 1999; Christensen et al., 1999; Devereux et al., 1999; Fausto, 1999; Anna et al., 2003), we are beginning to piece together the puzzle of the genetic basis of mouse liver carcinogenesis and regression. It is just a matter of time (and extensive analysis) that such critical pathways will be deciphered and applied in the treatment and prevention of cancer.
CONCLUSIONS
Progress has been made in the field of genomics of mouse neoplasia and promises are beginning to be fulfilled. Biological insights and pathways are being discovered that shed light on the processes of cancer initiation, differentiation, progression, and metastasis. In the mouse lung cancer model, gene expression profiles have been used to classify benign and malignant neoplasms as well as compare the expression profiles between lung cancer and normal lung, developing lung, and human lung cancer. Mammary gland and liver studies have identified alterations that are either independent or dependent on the underlying cause of cancer induction, whether it be genetically or chemically initiated. The finding of permanent gene expression alterations in adult mice after in utero exposure to arsenic has profound implications for identifying genetic risk to cancer development and understanding early genetic alterations of cancer. New genes are being discovered that have the potential to be novel oncogenes or tumor suppressor genes, biomarkers of progression or regression, or new therapeutic targets. It is clear that pathologists and toxicologists, in collaboration with molecular biologists, bioinformaticists, and other scientists are making great strides in the design, analysis, and interpretation of microarray data from cancer studies.
ACKNOWLEDGMENTS
We appreciate the assistance from Stella Sieber and Julie Foley from NIEHS, and Drs. Rick Hailey and Gary Boorman for their review and comments on the manuscript. We also appreciate the North Carolina Biotech Center (ARIG) and DuPont Center for Collaborative Research and Education for providing grants supporting the gene expression research in the chlordane tumor regression study.
REFERENCES
Anna, C. H., Iida, M., Sills, R. C., and Devereux, T. R. (2003). Expression of potential beta-catenin targets, cyclin D1, c-Jun, c-Myc, E-cadherin, and EGFR in chemically induced hepatocellular neoplasms from B6C3F1 mice. Toxicol Appl Pharmacol 190, 135–45.
Bank, U., Kruger, S., Langner, J., and Roessner, A. (2000). Review: peptidases and peptidase inhibitors in the pathogenesis of diseases. Disturbances in the ubiquitin-mediated proteolytic system. Protease-antiprotease imbalance in inflammatory reactions. Role of cathepsins in tumour progression. Adv Exp Med Biol 477, 349–78.
Bonner, A. E., Lemon, W. J., Devereux, T. R., Lubet, R. A., and You, M. (2004). Molecular profiling of mouse lung tumors: association with tumor progression, lung development, and human lung adenocarcinomas. Oncogene 23, 1166–76.
Bonner, A. E., Lemon, W. J., and You, M. (2003). Gene expression signatures identify novel regulatory pathways during murine lung development: implications for lung tumorigenesis. J Med Genet 40, 408–17.
Bratt, T. (2000). Lipocalins and cancer. Biochim Biophys Acta 1482, 318–26.
Bushel, P. R., Hamadeh, H., Bennett, L., Sieber, S., Martin, K., Nuwaysir, E. F., Johnson, K., Reynolds, K., Paules, R. S., and Afshari, C. A. (2001). MAPS: a microarray project system for gene expression experiment information and data validation. Bioinformatics 17, 564–5.
Challis, G. B., and Stam, H. J. (1990). The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol 29, 545–50.
Chen, X., Halberg, R. B., Ehrhardt, W. M., Torrealba, J., and Dove, W. F. (2003). Clusterin as a biomarker in murine and human intestinal neoplasia. Proc Natl Acad Sci USA 100, 9530–5.
Chiaverotti, T., Carabeo, R., and Drinkwater, N. (1999). Genetic and hormonal regulation of murine hepatocarcinogenesis. Prog Exp Tumor Res 35, 131– 42.
Chien, R. N., Chen, T. J., and Liaw, Y. F. (1992). Spontaneous regression of hepatocellular carcinoma. Am J Gastroenterol 87, 903–5.
Christensen, J. G., Romach, E. H., Healy, L. N., Gonzales, A. J., Anderson, S. P., Malarkey, D. E., Corton, J. C., Fox, T. R., Cattley, R. C., and Goldsworthy, T. L. (1999). Altered bcl-2 family expression during non-genotoxic hepatocarcinogenesis in mice. Carcinogenesis 20, 1583–90.
Currier, A. R., Sabla, G., Locaputo, S., Melin-Aldana, H., Degen, J. L., and Bezerra, J. A. (2003). Plasminogen directs the pleiotropic effects of uPA in liver injury and repair. Am J Physiol Gastrointest Liver Physiol 284, G508–15.
Desai, K. V., Kavanaugh, C. J., Calvo, A., and Green, J. E. (2002a). Chipping away at breast cancer: insights from microarray studies of human and mouse mammary cancer. Endocr Relat Cancer 9, 207–20.
Desai, K. V., Xiao, N., Wang, W., Gangi, L., Greene, J., Powell, J. I., Dickson, R., Furth, P., Hunter, K., Kucherlapati, R., Simon, R., Liu, E. T., and Green, J. E. (2002b). Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci USA 99, 6967– 72.
Desbois-Mouthon, C., Blivet-Van Eggelpoel, M. J., Beurel, E., Boissan, M., Delelo, R., Cadoret, A., and Capeau, J. (2002). Dysregulation of glycogen synthase kinase-3beta signaling in hepatocellular carcinoma cells. Hepatology 36, 1528–36.
Devereux, T. R., Anna, C. H., Foley, J. F., White, C. M., Sills, R. C., and Barrett, J. C. (1999). Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene 18, 4726–33.
Diez-Fernandez, C., Sanz, N., Alvarez, A. M., Wolf, A., and Cascales, M. (1998). The effect of non-genotoxic carcinogens, phenobarbital and clofibrate, on the relationship between reactive oxygen species, antioxidant enzyme expression and apoptosis. Carcinogenesis 19, 1715–22.
Emami, S., Le Floch, N., Bruyneel, E., Thim, L., May, F., Westley, B., Rio, M., Mareel, M., and Gespach, C. (2001). Induction of scattering and cellular invasion by trefoil peptides in src- and RhoA-transformed kidney and colonic epithelial cells. FASEB J 15, 351–61.
Emerson, Q. B., Nachtnebel, K. L., Penkava, R. R., and Rothenberg, J. (1980). Oral-contraceptive-associated liver tumours. Lancet 1, 1251.
Fan, C. Y., Pan, J., Chu, R., Lee, D., Kluckman, K. D., Usuda, N., Singh, I., Yeldandi, A. V., Rao, M. S., Maeda, N., and Reddy, J. K. (1996). Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem 271, 24698–710.
Fausto, N. (1999). Mouse liver tumorigenesis: models, mechanisms, and relevance to human disease. Semin Liver Dis 19, 243–52.
Fremond, B., Jouan, H., Sameh, A. H., Le Gall, E., Bergeron, C., Manac’h, A., Gruel, Y., and Babut, J. M. (1987). Tumors of the liver secondary to androgen therapy. Apropos of 2 cases in children. Chir Pediatr 28, 97–101.
Furukawa, T., Adachi, Y., Fujisawa, J., Kambe, T., Yamaguchi-Iwai, Y., Sasaki, R., Kuwahara, J., Ikehara, S., Tokunaga, R., and Taketani, S. (2001). Involvement of PLAGL2 in activation of iron deficient- and hypoxia-induced gene expression in mouse cell lines. Oncogene 20, 4718–27.
Gaffey, M. J., Joyce, J. P., Carlson, G. S., and Esteban, J. M. (1990). Spontaneous regression of hepatocellular carcinoma. Cancer 65, 2279–2783. Gong, Y., Cui, L., and Minuk, G. Y. (2000). The expression of insulin-like growth factor binding proteins in human hepatocellular carcinoma. Mol Cell Biochem 207, 101–4.
Gotoh, M., Sakamoto, M., Kanetaka, K., Chuuma, M., and Hirohashi, S. (2002). Overexpression of osteopontin in hepatocellular carcinoma. Pathol Int 52, 19–24.
Grasl-Kraupp, B., Ruttkay-Nedecky, B., Mullauer, L., Taper, H., Huber, W., Bursch, W., and Schulte-Hermann, R. (1997). Inherent increase of apoptosis in liver tumors: implications for carcinogenesis and tumor regression. Hepatology 25, 906–12.
Graveel, C. R., Harkins-Perry, S. R., Acevedo, L. G., and Farnham, P. J. (2003). Identification and characterization of CRG-L2, a new marker for liver tumor development. Oncogene 22, 1730–6.
Graveel, C. R., Jatkoe, T., Madore, S. J., Holt, A. L., and Farnham, P. J. (2001). Expression profiling and identification of novel genes in hepatocellular carcinomas. Oncogene 20, 2704–12.
Greaves, P., Irisarri, E., and Monro, A. M. (1986). Hepatic foci of cellular and enzymatic alteration and nodules in rats treated with clofibrate or diethylnitrosamine followed by phenobarbital: their rate of onset and their reversibility. J Natl Cancer Inst 76, 475–84.
Green, J. E., Desai, K., Ye, Y., Kavanaugh, C., Calvo, A., and Huh, J. I. (2004). Genomic approaches to understanding mammary tumor progression in transgenic mice and responses to therapy. Clin Cancer Res 10, 385S–90S.
Grisham, J. W. (1997). Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis 18, 59–81.
Huang, D. J., and Chen, H. C. (2004). Effects of chlordane and lindane on testosterone and vitellogenin levels in green neon shrimp (Neocaridina denticulata). Int J Toxicol 23, 91–5.
Iida, M., Anna, C. H., Hartis, J., Bruno, M., Wetmore, B., Dubin, J. R., Sieber, S., Bennett, L., Cunningham, M. L., Paules, R. S., Tomer, K. B., Houle, C. D., Merrick, A. B., Sills, R. C., and Devereux, T. R. (2003). Changes in global gene and protein expression during early mouse liver carcinogenesis induced by non-genotoxic model carcinogens oxazepam and Wyeth-14,643. Carcinogenesis 24, 757–70.
Ikeda, M., Okada, S., Ueno, H., Okusaka, T., and Kuriyama, H. (2001). Spontaneous regression of hepatocellular carcinoma with multiple lung metastases: a case report. Jpn J Clin Oncol 31, 454–8.
Kavanaugh, C., and Green, J. E. (2003). The use of genetically altered mice for breast cancer prevention studies. J Nutr 133, 2404S–2409S.
Koblinski, J. E., Ahram, M., and Sloane, B. F. (2000). Unraveling the role of proteases in cancer. Clin Chim Acta 291, 113–35.
Krepela, E. (2001). Cysteine proteinases in tumor cell growth and apoptosis. Neoplasma 48, 332–49.
Lam, K. C., Ho, J. C., and Yeung, R. T. (1982). Spontaneous regression of hepatocellular carcinoma: a case study. Cancer 50, 332–6.
Lee, P. D., Giudice, L. C., Conover, C. A., and Powell, D. R. (1997). Insulin-like growth factor binding protein-1: recent findings and new directions. Proc Soc Exp Biol Med 216, 319–57.
Leis, H., Segrelles, C., Ruiz, S., Santos, M., and Paramio, J. M. (2002). Expression, localization, and activity of glycogen synthase kinase 3beta during mouse skin tumorigenesis. Mol Carcinog 35, 180–5.
Lin, L., Wang, Y., Bergman, G., Kelloff, G. J., Lubet, R. A., and You, M. (2001). Detection of differentially expressed genes in mouse lung adenocarcinomas. Exp Lung Res 27, 217–29.
Liu, J., Xie, Y., Ward, J. M., Diwan, B. A., and Waalkes, M. P. (2004). Toxicogenomic analysis of aberrant gene expression in liver tumors and nontumorous livers of adult mice exposed in utero to inorganic arsenic. Toxicol Sci 77, 249–57.
Malarkey, D. E., Devereux, T. R., Dinse, G. E., Mann, P. C., and Maronpot, R. R. (1995). Hepatocarcinogenicity of chlordane in B6C3F1 and B6D2F1 male mice: evidence for regression in B6C3F1 mice and carcinogenesis independent of ras proto-oncogene activation. Carcinogenesis 16, 2617– 25. Malarkey, D. E., Johnson, K., Ryan, L., Boorman, G., and Maronpot, R. R. (2005). New Insights into Functional Aspects of Liver Morphology. Toxicologic Pathology 33, 27–34.
Maronpot, R. R., Fox, T., Malarkey, D. E., and Goldsworthy, T. L. (1995). Mutations in the ras proto-oncogene: clues to etiology and molecular pathogenesis of mouse liver tumors. Toxicology 101, 125–56.
Marsman, D. S., and Popp, J. A. (1994). Biological potential of basophilic hepatocellular foci and hepatic adenoma induced by the peroxisome proliferator, Wy-14,643. Carcinogenesis 15, 111–7.
Masui, T., Nakanishi, H., Inada, K., Imai, T., Mizoguchi, Y., Yada, H., Futakuchi, M., Shirai, T., and Tatematsu, M. (1997). Highly metastatic hepatocellular carcinomas induced in male F344 rats treated with N-nitrosomorpholine in combination with other hepatocarcinogens show a high incidence of p53 gene mutations along with altered mRNA expression of tumor-related genes. Cancer Lett 112, 33–45.
May, F. E., and Westley, B. R. (1997a). Expression of human intestinal trefoil factor in malignant cells and its regulation by oestrogen in breast cancer cells. J Pathol 182, 404–13.
May, F. E., and Westley, B. R. (1997b). Trefoil proteins: their role in normal and malignant cells. J Pathol 183, 4–7.
McCaughan, G. W., Bilous, M. J., and Gallagher, N. D. (1985). Long-term survival with tumor regression in androgen-induced liver tumors. Cancer 56, 2622–6.
Meyer, K., Lee, J. S., Dyck, P. A., Cao, W. Q., Rao, M. S., Thorgeirsson, S. S., and Reddy, J. K. (2003). Molecular profiling of hepatocellular carcinomas developing spontaneously in acyl-CoA oxidase deficient mice: comparison with liver tumors induced in wild-type mice by a peroxisome proliferator and a genotoxic carcinogen. Carcinogenesis 24, 975–84.
Nakae, J., Biggs, W. H., 3rd, Kitamura, T., Cavenee, W. K., Wright, C. V., Arden, K. C., and Accili, D. (2002). Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32, 245–53.
Nasu, K., Kai, K., Fujisawa, K., Takai, N., Nishida, Y., and Miyakawa, I. (2001). Expression of cathepsin L in normal endometrium and endometrial cancer. Eur J Obstet Gynecol Reprod Biol 99, 102–5.
Navab, R., Mort, J. S., and Brodt, P. (1997). Inhibition of carcinoma cell invasion and liver metastases formation by the cysteine proteinase inhibitor E-64. Clin Exp Metastasis 15, 121–9.
Pan, H. W., Ou, Y. H., Peng, S. Y., Liu, S. H., Lai, P. L., Lee, P. H., Sheu, J. C., Chen, C. L., and Hsu, H. C. (2003). Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer 98, 119–27.
Papac, R. J. (1998). Spontaneous regression of cancer: possible mechanisms. In Vivo 12, 571–8.
Price, J. A., Kovach, S. J., Johnson, T., Koniaris, L. G., Cahill, P. A., Sitzmann, J. V., and McKillop, I. H. (2002). Insulin-like growth factor I is a comitogen for hepatocyte growth factor in a rat model of hepatocellular carcinoma. Hepatology 36, 1089–97.
Qin, L. X., and Tang, Z. Y. (2004). Recent progress in predictive biomarkers for metastatic recurrence of human hepatocellular carcinoma: a review of the literature. J Cancer Res Clin Oncol 130, 497–513.
Scharf, J. G., Dombrowski, F., and Ramadori, G. (2001). The IGF axis and hepatocarcinogenesis. Mol Pathol 54, 138–44.
Scharf, J. G., Ramadori, G., and Dombrowski, F. (2000). Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab Invest 80, 1399–411.
Schneider, P., MacKay, F., Steiner, V., Hofmann, K., Bodmer, J. L., Holler, N., Ambrose, C., Lawton, P., Bixler, S., Acha-Orbea, H., Valmori, D., Romero, P., Werner-Favre, C., Zubler, R. H., Browning, J. L., and Tschopp, J. (1999). BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 189, 1747–56.
Seth, P., Porter, D., Lahti-Domenici, J., Geng, Y., Richardson, A., and Polyak, K. (2002). Cellular and molecular targets of estrogen in normal human breast tissue. Cancer Res 62, 4540–4.
Shackel, N. A., Gorrell, M. D., and McCaughan, G. W. (2002). Gene array analysis and the liver. Hepatology 36, 1313–25.
Simpson, P. T., Shoker, B. S., Barraclough, R., Halliwell, N., Rudland, P. S., Sibson, D. R., and Davies, M. P. (2003). Examination of tumour histopathology and gene expression in a neu/S100A4 transgenic model of metastatic breast cancer. Int J Exp Pathol 84, 173–84.
Steinbrecher, U. P., Lisbona, R., Huang, S. N., and Mishkin, S. (1981). Complete regression of hepatocellular adenoma after withdrawal of oral contraceptives. Dig Dis Sci 26, 1045–1050.
Takeda, Y., Togashi, H., Shinzawa, H., Miyano, S., Ishii, R., Karasawa, T., Saito, T., Saito, K., Haga, H., Matsuo, T., Aoki, M., Mitsuhashi, H., Watanabe, H., and Takahashi, T. (2000). Spontaneous regression of hepatocellular carcinoma and review of literature. J Gastroenterol Hepatol 15, 1079– 86.
Tang, Z. Y., Ye, S. L., Liu, Y. K., Qin, L. X., Sun, H. C., Ye, Q. H., Wang, L., Zhou, J., Qiu, S. J., Li, Y., Ji, X. N., Liu, H., Xia, J. L., Wu, Z. Q., Fan, J., Ma, Z. C., Zhou, X. D., Lin, Z. Y., and Liu, K. D. (2004). A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol 130, 187–96.
van Es, J. H., Kirkpatrick, C., van de Wetering, M., Molenaar, M., Miles, A., Kuipers, J., Destree, O., Peifer, M., and Clevers, H. (1999). Identification of APC2, a homologue of the adenomatous polyposis coli tumour suppressor. Curr Biol 9, 105–8.
van Loo, G., van Gurp, M., Depuydt, B., Srinivasula, S. M., Rodriguez, I., Alnemri, E. S., Gevaert, K., Vandekerckhove, J., Declercq, W., and Vandenabeele, P. (2002). The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 9, 20–6.
Wright, N. A., Hoffmann, W., Otto, W. R., Rio, M. C., and Thim, L. (1997). Rolling in the clover: trefoil factor family (TFF)-domain peptides, cell migration and cancer. FEBS Lett 408, 121–3.
Xu, S., and Venge, P. (2000). Lipocalins as biochemical markers of disease. Biochim Biophys Acta 1482, 298–307.
Yakar, S., Wu, Y., Setser, J., and Rosen, C. J. (2002). The role of circulating IGF-I: lessons from human and animal models. Endocrine 19, 239–48.
Yang, C., and Chen, S. (1999). Two organochlorine pesticides, toxaphene and chlordane, are antagonists for estrogen-related receptor alpha-1 orphan receptor. Cancer Res 59, 4519–24.
Yao, R., Wang, Y., Lubet, R. A., and You, M. (2002). Differentially expressed genes associated with mouse lung tumor progression. Oncogene 21, 5814– 21.