An embryo-fetal survival and development study was conducted to augment the toxicity database for alpha-glycosyl isoquercitrin (AGIQ), a generally recognized as safe (GRAS) additive and flavor in food and beverages. In Phase I, 24 naturally mated New Zealand white (NZW) female rabbits per group were administered AGIQ by oral gavage at 0, 250, 500, or 1000 mg/kg/day once daily during gestation days 6–28, followed by necropsy. There was no evidence of maternal or fetal toxicity except for equivocal findings of unilateral absent kidney and ureter in one and two unrelated fetuses at 500 and 1000 mg/kg/day, respectively. To more thoroughly assess fetal kidney/ureter development, in Phase II groups of time mated NZW rabbits were administered AGIQ at 0, 500, or 1000 mg/kg/day, under the same conditions as Phase I. No occurrences of absent kidney/ureter were noted in the AGIQ-treated Phase II dams or fetuses; although, one control fetus had unilateral missing kidney/ureter. Given the lack of reproducibility following treatment with AGIQ in Phase II using 48 animals per group, the missing kidney/ureter observations in Phase I were considered unrelated to treatment. Since oral gavage administration of AGIQ to pregnant female NZW rabbits at dose levels of 250, 500, or 1000 mg/kg/day was well-tolerated with no adverse treatment-related effects on the maternal animal, pregnancy, or the developing conceptus, the no-observed-adverse-effect-level (NOAEL) for maternal toxicity and embryo-fetal survival, growth, and development was 1000 mg/kg/day.
Introduction
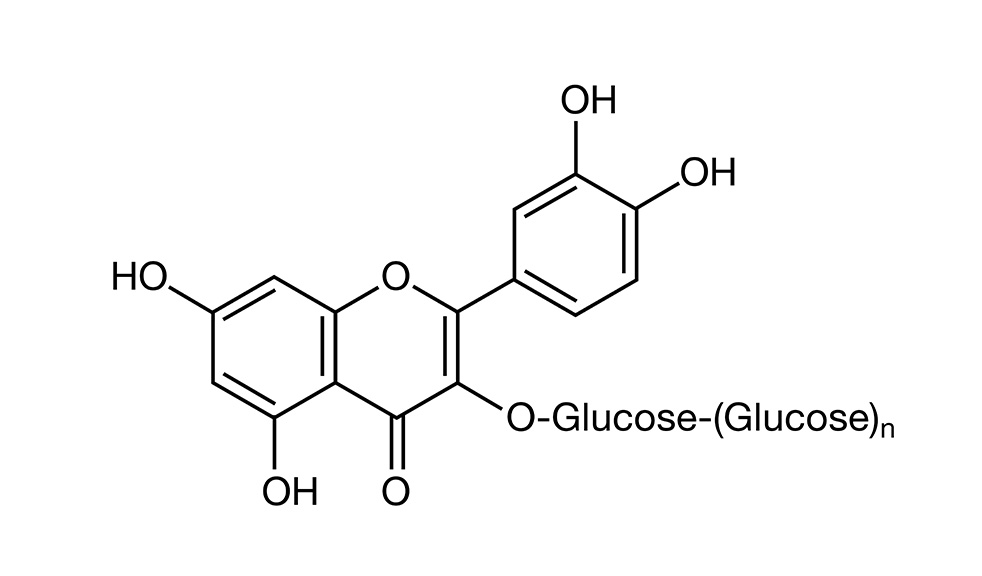
Alpha-glycosyl isoquercitrin (AGIQ) (Figure 1) is an enzymatically modified form of the natural flavonol isoquercitrin (quercetin-3-O-ß D-glucoside), derived from rutin and used in Japan as an additive or flavor ingredient in various beverages and foods. Although quercetin and its glycosides have demonstrated anti-inflammatory, pain-reducing, and cardioprotective properties1–6 and therefore have been promoted as antioxidant dietary supplements to consumers, their poor miscibility in water and limited absorption have hindered their broad application to the food and beverage industry.7 However, the enzymatic modification resulting in AGIQ has been shown to enhance the solubility and bioavailability of isoquercitrin.8,9
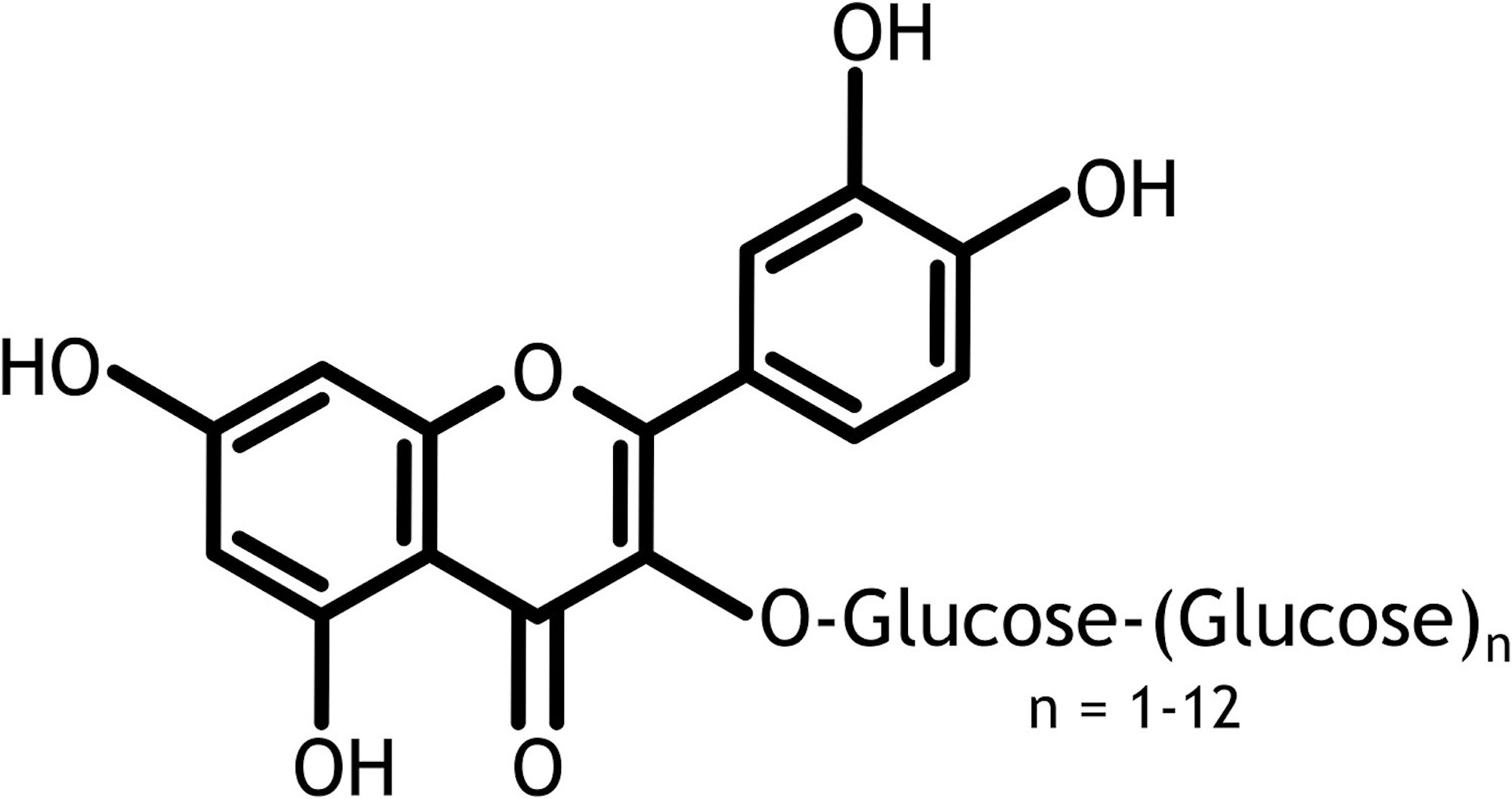
Figure 1. Chemical formula of alpha-glycosyl isoquercitrin.
Previous toxicity assessments have shown that AGIQ is safe, non-carcinogenic, and non-genotoxic,5,7,10–13 but some of the earlier studies used incompletely characterized AGIQ and/or did not adhere to current Good Laboratory Practice (GLP).6,10,12,13 Due to the current effort to expand the use of AGIQ in the consumer food and beverage industry, recent GLP-compliant studies of high-purity AGIQ (including comprehensive genotoxic assessment,7 10-day and 4-week studies in preweaning Göttingen minipigs,14 and a 90-day study in rats)5 were conducted to augment the older toxicity database. These studies have generally confirmed the safety profile of AGIQ.
The current study using highly purified AGIQ and New Zealand White (NZW) rabbits was conducted to assess the potential effects of the compound on embryo-fetal development, growth, and survival in a non-rodent species for registration purposes. The objective of the initial dose-response phase of the study (Phase I) was to assess the potential of AGIQ to induce prenatal developmental toxicity after maternal exposure via oral gavage during the critical period of organogenesis and fetal development. A subsequent investigational phase (Phase II) was conducted using 48 animals in each group to verify equivocal Phase I findings of unilateral absent fetal kidney/ureter in one and two (unrelated) fetuses at the mid and high dose levels, respectively. This study provides a base of non-rodent developmental toxicity data obtained in accordance with current developmental toxicity testing guidelines (Phase I)15–17 and GLP (both phases)18–21 for human risk assessment of orally administered AGIQ.
Materials and methods
Test article
AGIQ (>97% pure, 0.13% quercetin, lot no. 170727) was supplied as a yellow to yellow-orange powder by San-Ei Gen F.F.I., Inc., Osaka, Japan.
Dosing formulations were prepared by dissolving AGIQ in purified water at the target dose concentrations of 25, 50, and 100 mg/mL. Representative samples of each formulation concentration prepared for administration during each phase were analyzed on two occasions for achieved concentration of AGIQ. A previous GLP-compliant validation study (Envigo Study No. NL85VW, 21 June 2018; data not presented) found that solutions of AGIQ in purified water at concentrations ranging from 1 to 200 mg/mL were homogeneous and stable for 1 day when stored at ambient temperature (15–25°C) and 15 days when stored refrigerated (2–8°C). Based on these results, the formulations in this study were prepared daily, stored at ambient temperature until use, and administered within 4 h of preparation.
Animal husbandry and mating
Female NZW rabbits (obtained from Envigo RMS UK) were used for this research. For Phase I, 96 sexually mature, virgin females uniquely identified by an ear tag were supplied (88 assigned to study and 8 serving as potential replacements). For Phase II, 144 time-mated females (all assigned to study) were supplied in four deliveries on GD 1 after mating at the supplier. Phase II animals were uniquely identified by a microchip inserted shortly after arrival. Each cage label was color-coded according to group and was numbered uniquely with cage number, study number, and the identity of the occupant.
The rabbits were allowed a 19-day (Phase I) or 5-day (Phase II) period of acclimation to the facility conditions prior to allocation to the study (mating in Phase I [GD 0] or day of arrival [GD 1] in Phase II). Female rabbits were cohabited with stock NZW bucks of established fertility at the performing laboratory (Phase I) or the supplier (Phase II). Males and females were not closely related. Phase I females were assigned to group and cage position in the sequence of observed natural mating (females mating on the same day were evenly distributed among the groups) and Phase II females were randomly assigned to group and cage position, evenly distributed among the groups. After mating, each female was injected intravenously with 25 i.u. luteinizing hormone. The day of observed mating was designated GD 0. Allocation was controlled to prevent any stock male from providing more than one mated female in each treated group and to prevent more than one sibling female in each group, where possible.
The animals were 19–23 (Phase I) or 16–20 (Phase II) weeks of age at the start of the study (GD 0/1). The basal diet, Teklad 2930 Diet, was restricted to 150 g/animal/day during acclimation up to 1 week prior to the onset of mating (Phase I) and 200 g/animal/day thereafter (Phase I and II). In addition to the basal diet, a small supplement of autoclaved hay was given on a daily basis to promote gastric motility and a small amount of chopped fresh vegetables were given twice weekly. Consumption of hay and vegetables were monitored qualitatively but not quantitatively. The animals were allowed unrestricted access to potable drinking water from the public supply, supplied via polycarbonate water bottles equipped with sipper tubes and supplementary water bowls in each cage, throughout the study. Water bottles/bowls were changed at appropriate intervals. All animals were housed individually (except while paired for mating (Phase I) in suspended cages fitted with perforated floor panels and plastic resting platforms. Undertrays lined with absorbent paper were changed at least three times per week. All animals were maintained on a 14-hour daily photoperiod (10 hours dark) at an environmental temperature of 15–21°C and relative humidity of 45–70%. Environmental enrichment for each animal consisted of an Aspen chew block (soft white untreated wood block), a stainless-steel key ring attached to the cage, and nesting paper placed into each cage starting at post-mating GD 20 to allow expression of nesting behavior.
Phase I and II were conducted in an AAALAC accredited facility (Covance Laboratories Limited, Eye, UK (Study JJ43CT)) in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012.22 The number of animals used was the minimum that was consistent with scientific integrity and regulatory acceptability, consideration having been given to the welfare of individual animals in terms of the number and extent of procedures to be carried out on each animal. The NZW rabbit was chosen as the test model based on the availability of Historical Control Data (HCD) in the performing laboratory and the acceptability of this species and strain to regulatory agencies.
Experimental design
This study was conducted in two phases (Table 1): (I) a regulatory guideline-compliant embryo-fetal survival and development study and (II) a follow-up confirmatory investigation of equivocal fetal morphological findings noted in Phase I.

In a preliminary dose range-finding study conducted in the same laboratory, administration of AGIQ to 6 females per group was well-tolerated at 250, 500, and 1000 mg/kg/day, but was not tolerated at 2000 mg/kg/day and this group of animals was terminated early on GD 21/22 due to marked body weight loss and persistently low food consumption in some females. The urine from all treated animals was colored yellow and stained the cage tray paper and animal fur yellow/orange/ brown and the amniotic fluid/sacs in 1/6 animals at 250 mg/kg/day and 1/6 animals at 500 mg/kg/day, but none at 1000 mg/kg/day, were colored yellow. The strong coloration was attributed to the color of AGIQ (yellow to yellow-orange powder). At 1000 mg/kg/day, overall body weight gain was low and food intake was marginally low. Reproductive parameters did not appear to be affected by treatment with AGIQ.
Based on these preliminary results, female rabbits in Phases I (22/group) and II (48/group) were administered AGIQ in purified water once daily during GD 6–28 (inclusive) at approximately the same time each day via oral gavage at a dose volume of 10 mL/kg (calculated from the most recently recorded scheduled body weight) using a suitably graduated cylinder and rubber catheter inserted via the mouth. Phase I dose levels were 0 (purified water vehicle), 250, 500, and 1000 mg/kg/day and Phase II dose levels were 0, 500, and 1000 mg/kg/day. Surviving females in each phase were euthanized on GD 29 for maternal and fetal examinations.
Maternal evaluation (Phases I and II)
Clinical observations were conducted daily during the acclimation period and at least twice daily during the study for evidence of ill-health or reaction to treatment. Detailed observations for signs associated with dosing were conducted daily during the treatment period prior to dosing, 1–2 hours after completion of dosing, and as late as possible in the working day. A detailed physical examination of each animal was also conducted on GD 0 (Phase I only), 1 (Phase II only), 6, 12, 18, 23, and 29 to monitor general health. Maternal body weights were collected during acclimation and on the day of mating (GD 0; Phase I), on arrival (GD 1; Phase II), and on GD 3 and 6–29 (daily). Food consumption was measured daily during the study. All surviving adult females were euthanized on GD 29 via an intravenous injection of sodium pentobarbitone and all viable fetuses were euthanized via a subcutaneous injection of sodium pentobarbitone. One control and two dosed rabbits were euthanized for welfare reasons and all were pregnant.
Detailed necropsies were conducted for Phase I females, including full macroscopic examination of tissues (including the kidney and ureters) and visual examination of all external features and orifices. Any abnormality in the appearance or size of any organ and tissue (external and cut surface) was recorded and the required tissue samples were preserved in appropriate fixative.
Postmortem examinations for Phase II females were limited to confirmation of pregnancy status and examination for the presence/absence of kidney/ureters and any abnormalities in these organs.
Reproductive assessment (Phases I and II)
For Phase I, the uterus of each dam was excised and gravid uterine weights (including cervix and ovaries) were obtained and recorded. For each ovary/uterine horn, corpora lutea were counted and the number and location of viable and nonviable fetuses, early and late resorptions, and total number of implantation sites were recorded. For apparently nonpregnant animals and apparently empty uterine horns, the absence or number of uterine implantation sites was confirmed by visual examination.
For Phase II females that survived to term, all live fetuses were examined only for the presence/absence of kidney/ureters and any abnormalities in these organs. Fetuses with macroscopic findings or abnormalities in these organs were fixed in neutral-buffered formalin and retained. Representative photographs were taken of at least one male and one female fetus from each group to document the renal/ureter relationship and any fetus(es) with renal/ureter abnormalities.
Fetal macropathology (Phase I)
All viable fetuses and placentae were dissected from the uterus, individually weighed, identified within the litter using a coding system based on their position in the uterus, examined externally with abnormalities recorded, sampled as appropriate, and retained in 10% neutral-buffered formalin fixative. All fetuses were subjected to a gross internal examination of the viscera of the neck, thorax, and abdominal cavities. The sex of each fetus was also recorded. Approximately one-half of the eviscerated fetuses were decapitated and the heads were initially stored in Bouin’s fluid. The remaining eviscerated fetuses and torsos were fixed in Industrial Methylated Spirit. Fetal heads fixed in Bouin’s fluid were subjected to freehand serial sections, which were examined for soft tissue abnormalities. The fetuses and torsos fixed in Industrial Methylated Spirit were processed and stained with Alizarin Red S, then assessed for skeletal development and abnormalities.
Findings from external, visceral, and skeletal examination of fetuses were classified, according to severity and incidence, as either major abnormalities, minor abnormalities, or variants. Major abnormalities are normally rare, definitely detrimental to normal subsequent development, and possibly lethal (e.g., partially open eyelids or absent kidney/ureter). Minor abnormalities are minor differences from normal that are detected relatively frequently, are considered to have little detrimental effect, and may be a transient stage in development (e.g., bipartite centrum or dilated ureter). Variants are alternative structures or stages of development occurring regularly in the control population (e.g., number of ribs and thoracolumbar vertebrae or incomplete ossification of fifth and sixth sternebrae).
Statistical analysis
All statistical analyses were conducted for minimum significance levels of 5% and 1%, comparing each AGIQ-treated group to the appropriate control group by phase and sex. Maternal and fetal developmental toxicity endpoints were analyzed using the maternal animal or the litter as the experimental unit, as appropriate. For Phase I reproductive assessment (i.e., corpora lutea, implantations, resorptions, litter size, live young, and pre- and post-implantation losses) and fetal, litter, and placental weight data, group mean values and standard deviations were calculated using individual litter mean values, as appropriate. Standard deviations were not calculated for derived data, such as levels of pre- and post-implantation loss, or for the incidence of resorbing fetuses where the distribution of these findings commonly does not conform to the normal statistical distribution.
Continuous data variables (mean maternal body weights, body weight changes, and food consumption), gravid uterine weight and adjusted body weight, corpora lutea, implantations, litter size, live young, and placental, litter, and fetal body weights were subjected to a parametric analysis if Bartlett’s test for variance homogeneity23 was not significant at the 1% level. For pretreatment data, analysis of variance was used to test for any group differences. Where this analysis was significant (p < 0.05), intergroup comparisons were made using t-tests, with the error mean square from the one-way analysis of variance. For all other comparisons the F1 approximate test was applied. This test was designed to detect significant departure from monotonicity of means when the main test for the comparison of the means is a parametric monotonic trend test, such as Williams’ test.24,25 The test statistic compared the mean square, NMS, for the deviations of the observed means from the maximum likelihood means, calculated under a constraint of monotonicity with the usual error mean square, EMS. The null hypothesis was that the true means were monotonically ordered. The test statistic was F1 = NMS/EMS, which can be compared with standard tables of the F-distribution with 1 and error degrees of freedom. If the F1 approximate test for monotonicity of dose-response was not significant at the 1% level, Williams’ test for a monotonic trend was applied. If the F1 approximate test was significant, suggesting that the dose-response was not monotone, Dunnett’s test26,27 was performed instead.
For the data variables described above, a non-parametric analysis was performed if Bartlett’s test was still significant at the 1% level following both logarithmic and square-root transformations. For pretreatment data, the Kruskal–Wallis test28,29 was used to test for any group differences. Where this analysis was significant (p < 0.05), intergroup comparisons using Wilcoxon rank sum tests30 were made. For all other comparisons, the H1 approximate test, the non-parametric equivalent of the F1 test described above, was applied. This test was designed to be used when the main test for comparison of the means is a non-parametric monotonic trend test, such as Shirley’s test (Shirley 1977).31 The test statistic compared the non-monotonicity sums of squares, NRSS, for the deviations of the observed mean ranks from the maximum likelihood mean ranks with the non-parametric equivalent of the error sums of squares, ERSS = N(N + 1)/12. The test statistic was H1 = NRSS/ERSS, which can be compared to standard tables of the χ2-distribution with 1 degree of freedom. If the H1 approximate test for monotonicity of dose-response was not significant at the 1% level, Shirley’s test for a monotonic trend was applied. If the H1 approximate test was significant, suggesting that the dose-response was not monotone, Steel’s test32 was performed instead.
For litter size, litter survival indices, and gravid uterine, placental, litter, and fetal body weight data (Phase I only), if 75% of the data (across all groups) were the same value, for example c, Fisher’s exact tests33 were performed. Treatment groups were compared using pairwise comparisons of each dose group against the control both for 1) values <c versus values ≥c and for 2) values ≤c versus values >c, as applicable.
Pre- and post-implantation loss and fetal sex ratio (Phase I only) were analyzed by a generalized mixed linear model with binomial errors, a logit link function, and litter as a random effect (Lipsitz 1991).34 Each treated group was compared to the control group using a Wald chi-square test. For pre-implantation loss (reflecting losses due to non-fertilization of ova and failure to implant), the numerator was number of corpora lutea minus number of implantations, and the denominator was number of corpora lutea. For post-implantation loss, the numerator was number of implantations minus number of live fetuses, and the denominator was number of implantations. For fetal sex ratio, the numerator was number of males and the denominator was number of live fetuses.
For resorptions (Phase I only), each treated group was compared to the control group by exact Wilcoxon rank sum test (Wilcoxon 1945).30
Results
Formulation analysis
The mean analyzed concentrations of AGIQ formulations prepared for Phases I and II were within 104% to 110% of the nominal concentrations, confirming the accuracy of the formulations. The differences from the mean values were within 5%, which confirmed precision of analysis.
Maternal effects (Phases I and II)
Maternal data are presented in Table 2 and Figures 2 and 3.
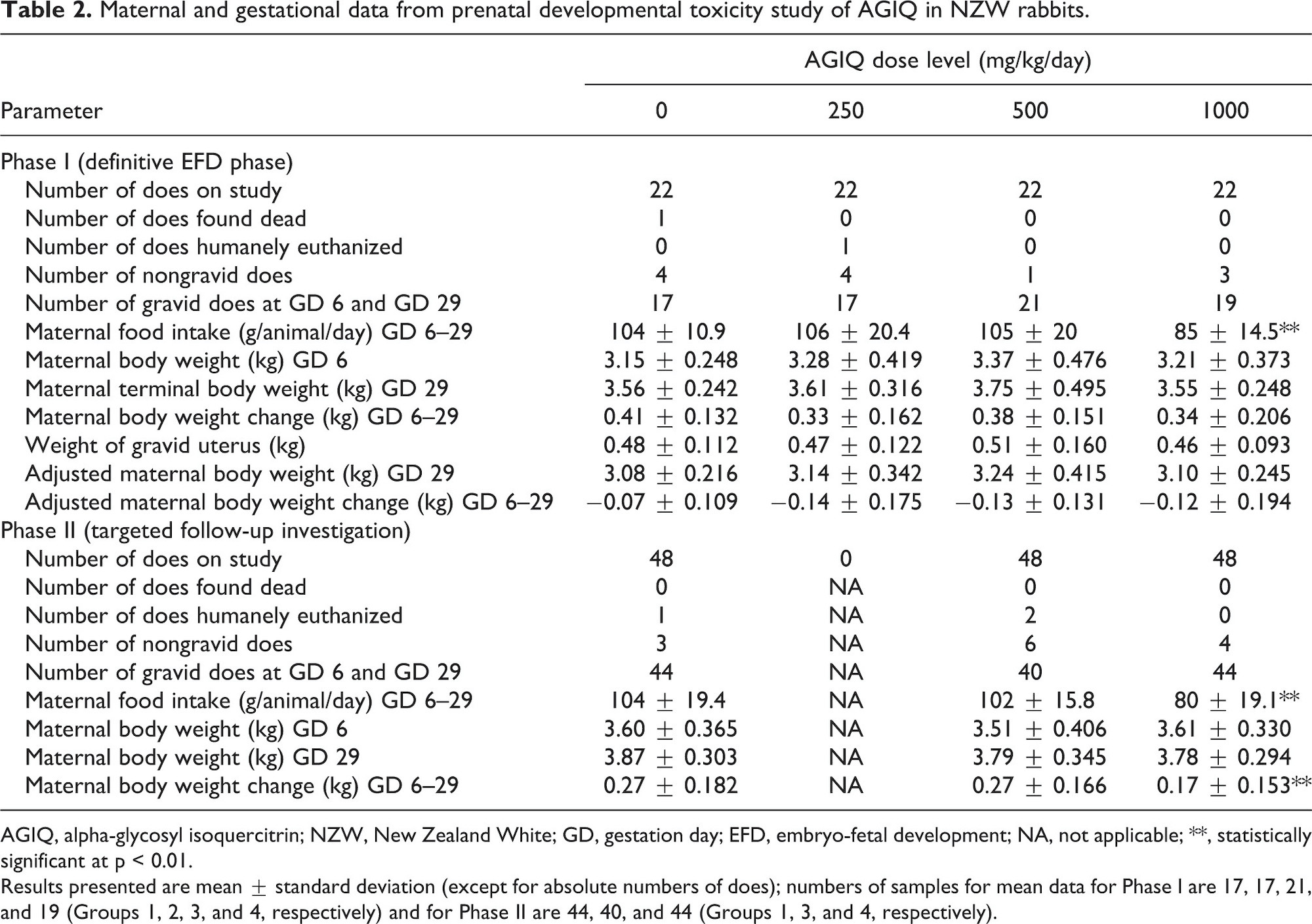
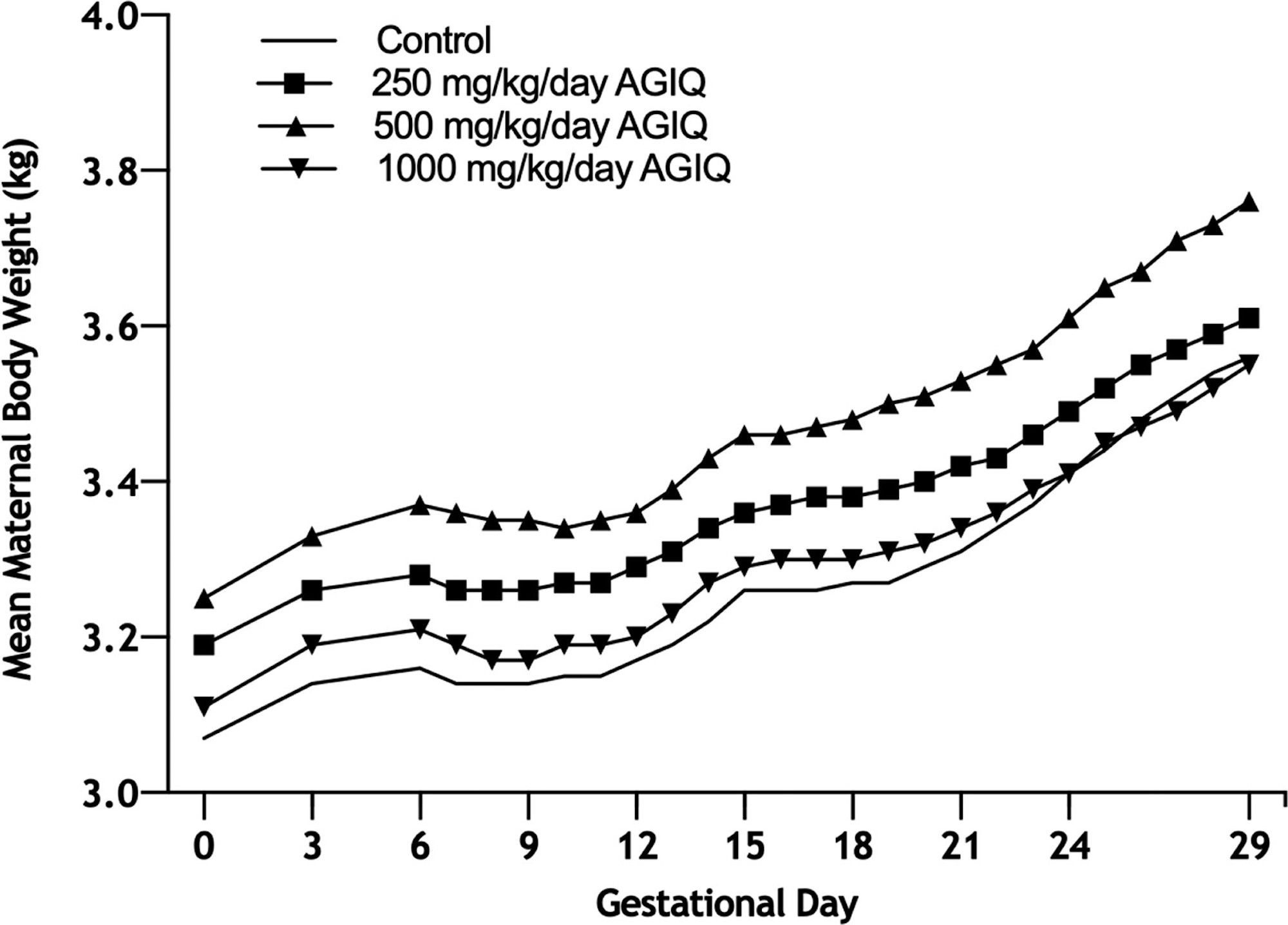
Figure 2. Growth curves of Phase I pregnant New Zealand White rabbits administered AGIQ at doses of 0, 250, 500, and 1000 mg/kg/day (N = 17–18, 17, 21, and 19, respectively) during gestational days (GD) 6–28, followed by necropsy and comprehensive fetal examinations on GD 29. AGIQ, alpha-glycosyl isoquercitrin. There were no statistically significant differences from control.
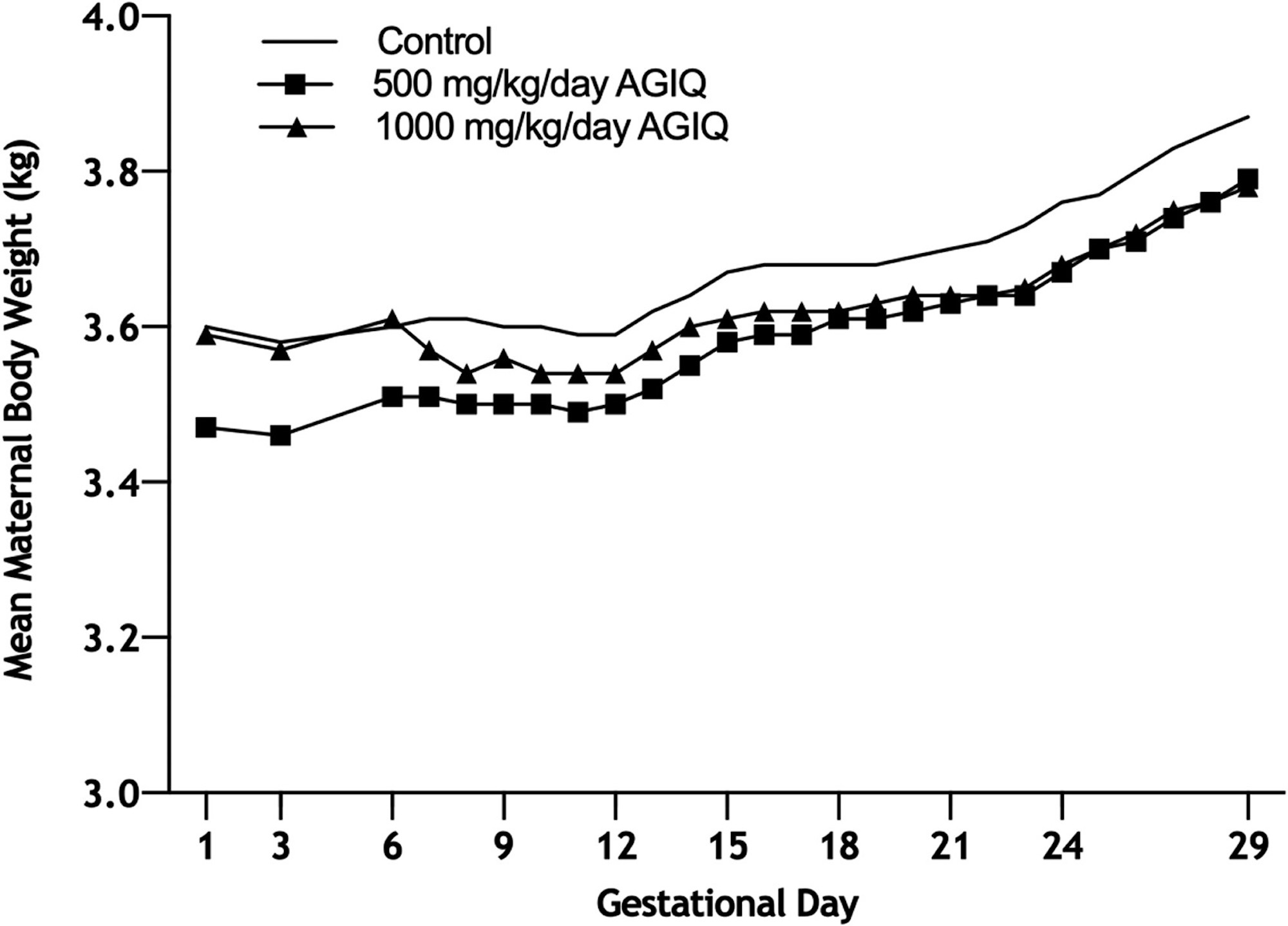
Figure 3. Growth curves of Phase II pregnant New Zealand White rabbits administered AGIQ at doses of 0, 500, and 1000 mg/kg/day (N = 44, 40, and 44, respectively) during gestational days (GD) 6–28, followed by examination of maternal and fetal kidneys/ureters on GD 29. AGIQ, alpha-glycosyl isoquercitrin. There were no statistically significant differences from control.
There was no effect on maternal survival or macropathology in either phase following AGIQ administration at 250 (Phase I only), 500, or 1000 mg/kg/day. There were no macroscopic findings related to AGIQ treatment at the postmortem examinations of maternal animals, other than some yellow staining of the fur in all treated groups (data not shown). All females in Phase I (22 adults each at 0, 250, 500, and 1000 mg/kg/day) and Phase II (48 adults each at 0, 500, and 1000 mg/kg/day) had macroscopically normal, paired kidneys and ureters, except for one Phase I female at 250 mg/kg/day that had cysts on the kidneys.
There were no adverse clinical signs associated with AGIQ dose administration at any dose level. However, maternal body weight gain at 1000 mg/kg/day in Phase II was consistently lower than body weight change at 500 mg/kg/day and in the controls. The only remarkable clinical finding in both phases was a high incidence, when compared with concurrent controls, of non-adverse yellow staining of the mouth, head, forepaws, hind paws, and tail in animals treated at 250, 500, or 1000 mg/kg/day (data not presented). This staining was attributed to the color of AGIQ.
Maternal food intake during GD 6–29 in Phases I and II was statistically significantly lower than controls at 1000 mg/kg/day (82% or 77% lower, respectively) and was unaffected by treatment at 250 (Phase I only) and 500 mg/kg/day. Maternal body weights in both phases and overall body weight gain during GD 6–29 at 250 (Phase I only), 500 (both phases), and 1000 (Phase I) mg/kg/day were unaffected by treatment. Overall body weight gain during GD 6–29 at 1000 mg/kg/day in Phase II was statistically significantly lower than control, but the difference was marginal and considered not to be biologically relevant or adverse. Gravid uterine weight, adjusted body weight (GD 29), and adjusted body weight change (GD 6–29) were unaffected by treatment at any dose level in Phase I.
Pregnancy outcome (Phase I)
Phase I litter data, including placental, litter, and fetal weights, are presented in Table 3.
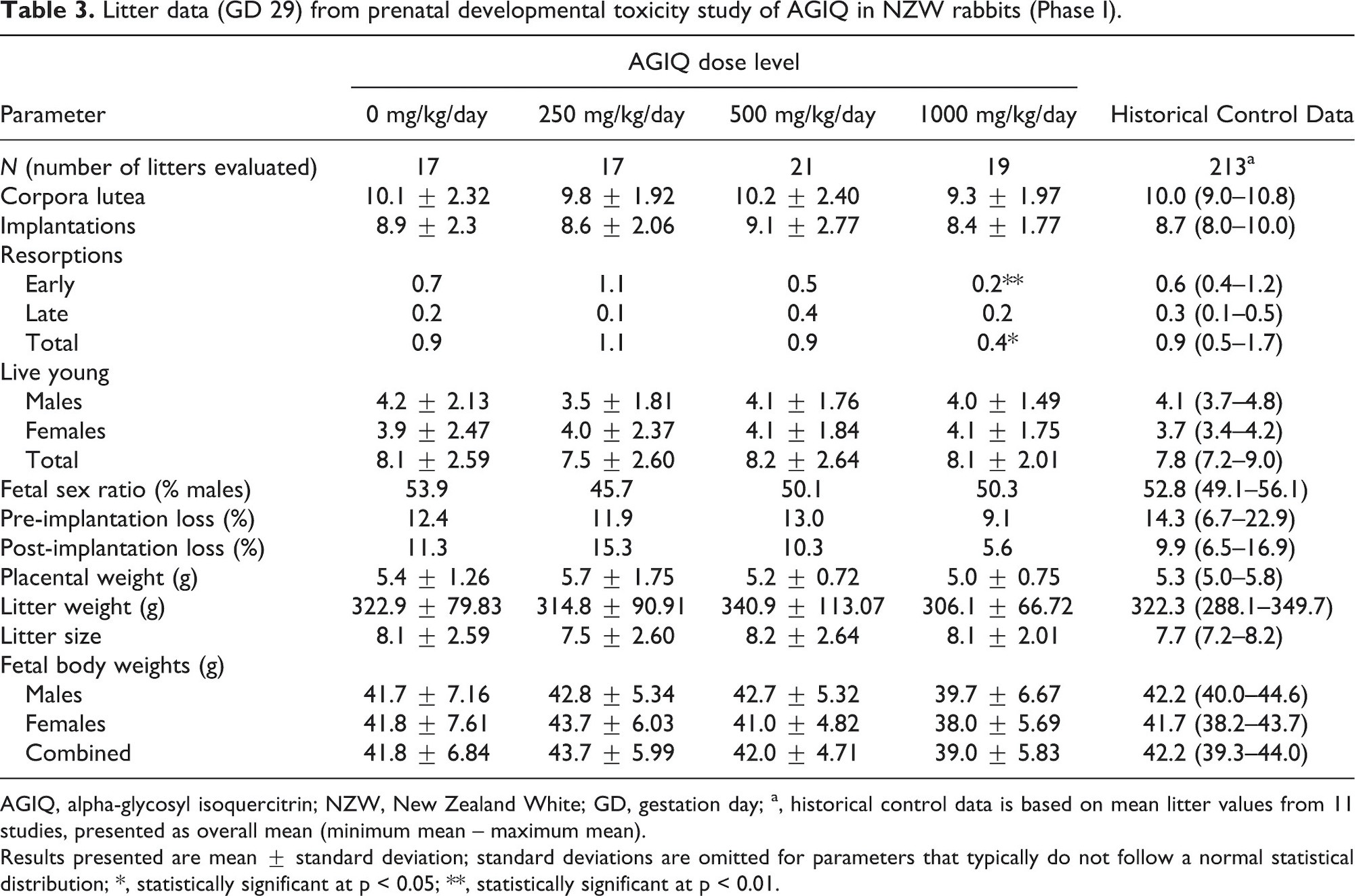
The mean numbers of implantations, implantation losses, resorptions, live young, fetal sex ratios, and placental and litter weights were similar to control values. Mean fetal weights at 1000 mg/kg/day were slightly lower than concurrent controls (not statistically significant) and the HCD range; this decrease is considered minor and indicative of a slight delay in fetal development that would resolve over time and is therefore considered non-adverse.
Fetal macropathology (Phase I)
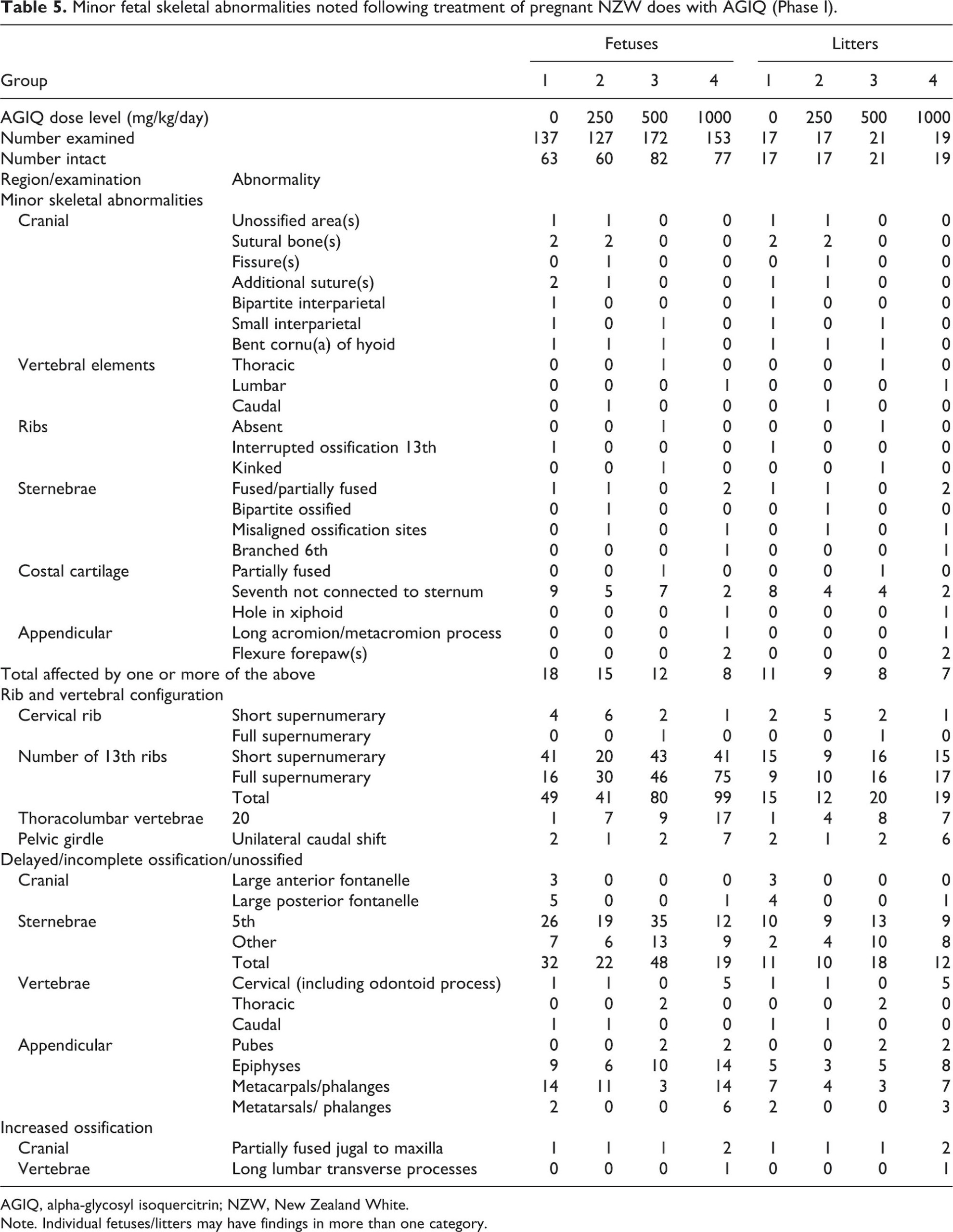
Fetal macropathology data for Phase I are presented in Table 4 (major abnormalities), Table 5 (minor skeletal abnormalities), and Table 6 (minor visceral abnormalities and necropsy findings).
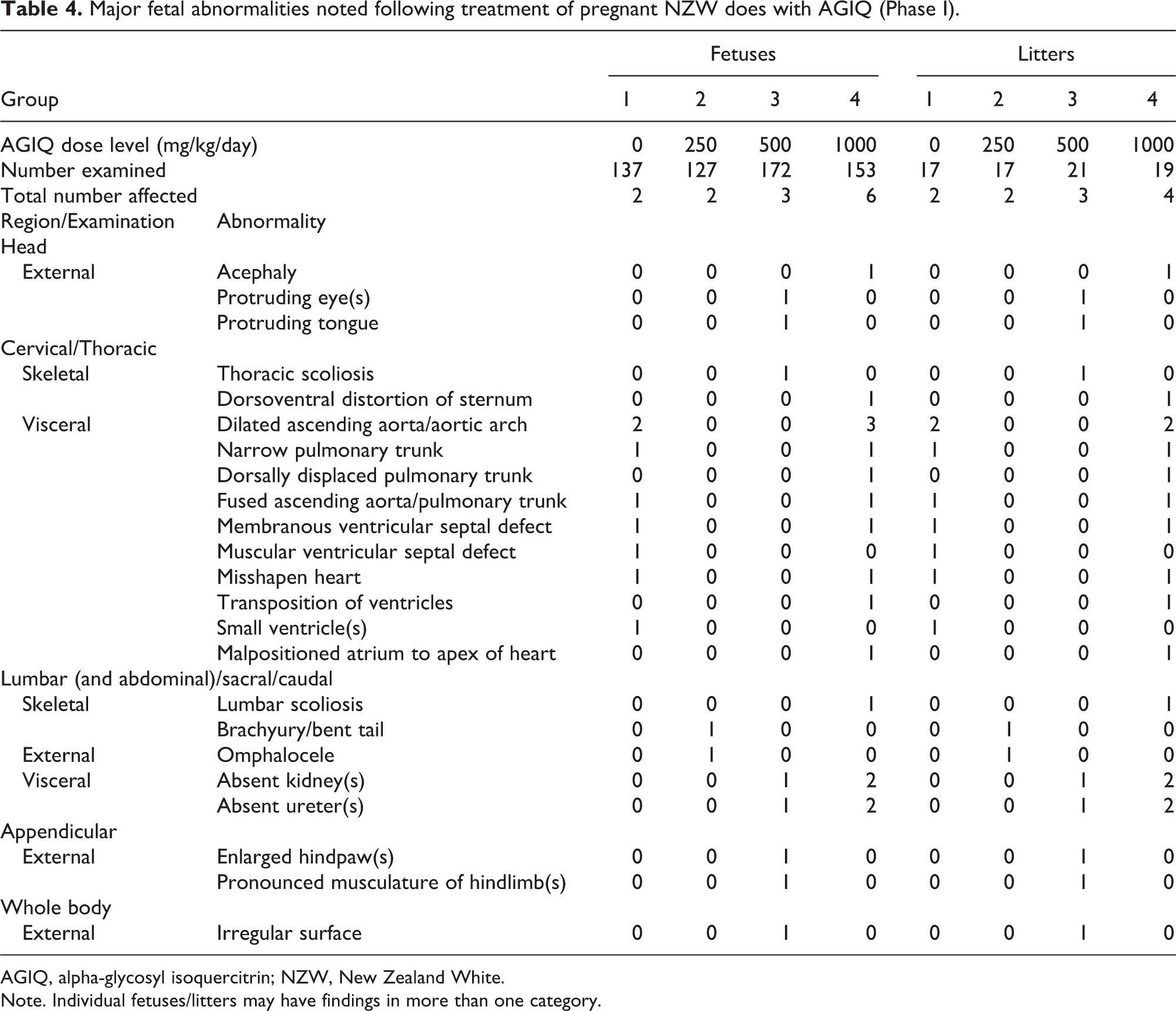
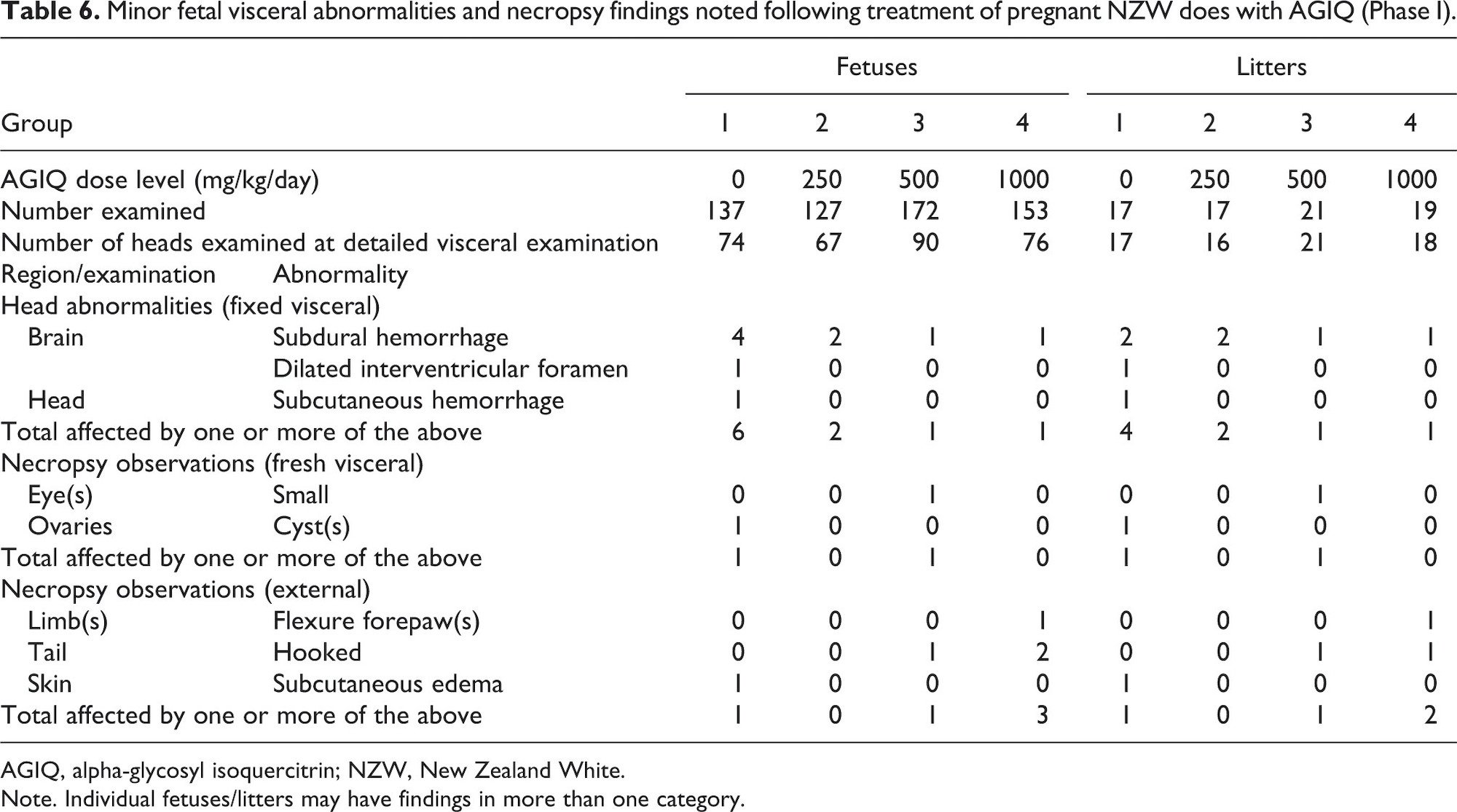
Oral administration of AGIQ to pregnant NZW rabbits did not adversely affect fetal external, visceral, or skeletal morphology.
Examination of 589 Phase I fetuses across four groups revealed that one fetus in the 500 mg/kg/day group and single fetuses in two separate litters in the 1000 mg/kg/day group had unilateral absent ureter and kidney, compared with one fetus in one litter noted in the performing laboratory’s HCD with this finding. Based on the rare nature of this finding, the fact that it matched or exceeded the historical control data incidence, and an apparent dose responsive occurrence, the absent kidney/ureter findings at 500 and 1000 mg/kg/day were considered potential treatment-related effects. However, the very low incidence of the findings (one or two fetuses per group) suggested that they were most likely spurious, spontaneous clusters. Therefore, a follow-up investigation in Phase II was conducted.
In Phase II, examination of 1028 fetuses across the control, 500, and 1000 mg/kg/day groups revealed that all fetuses had paired kidneys and ureters, with the exception of one control fetus that had a single kidney and ureter. Therefore, the findings of unilateral absent ureter and kidney at 500 and 1000 mg/kg/day in Phase I is considered normal background variability unrelated to AGIQ treatment.
Some minor skeletal abnormalities were evident at 1000 mg/kg/day (Phase I). There were increases in the incidence of full supernumerary 13th rib (exceeded the HCD range) and 20 thoracolumbar vertebrae and unilateral caudal shift of the pelvic girdle (both within the HCD range). The combination of these skeletal abnormalities indicated a slight shift in rib/vertebral configuration that was considered non-adverse. There was also a slight increase in the incidence of incompletely ossified cervical vertebral elements (within the HCD range) and epiphyses (exceeded the HCD range). These findings are indicative of a slight delay in fetal development that is considered non-adverse.
All other fetal external, visceral, and skeletal abnormalities that occurred in this study were not attributed to maternal AGIQ exposure because they were limited to single fetuses or litters, there was no dose-response relationship evident, and/or the findings had been previously observed in control NZW rabbit fetuses evaluated in the performing laboratory.
Discussion
In Phase I, oral administration of AGIQ to pregnant NZW rabbits at 250, 500, or 1000 mg/kg/day was well-tolerated, with no effect on reproductive parameters, specifically gravid uterine weight, number of implantations, implantation losses, resorptions, live young, the ratio of males to females, or on placental, litter, and fetal weights and litter size. Food consumption at 1000 mg/kg/day was marginally low at 82% of control throughout the treatment period; however, there was no resultant effect on maternal body weight or gravid uterine weight and therefore the lower food consumption is considered to be non-adverse. The cage tray papers of all animals in all treated groups were discolored/stained yellow, orange, and/or brown from urine and there was some yellow staining of the fur. This strong coloration was attributed to the color of AGIQ (yellow to yellow-orange powder) and is considered to be non-adverse. Macroscopic yellow coloration in previous AGIQ studies has not been associated with histopathological change.5,36
At 500 and 1000 mg/kg/day, the major abnormality of unilateral absent kidney and ureter was evident in 1/172 examined fetuses at 500 mg/kg/day and 2/153 examined fetuses (in two unrelated litters) at 1000 mg/kg/day, compared with 0/127 and 0/137 in the 250 mg/kg/day and concurrent control group, respectively. Because the incidence of unilateral kidney in the 1000 mg/kg/day group exceeded the upper range of the performing laboratory missing unilateral kidney (viz., 1/1315), the possibility of a treatment-related effect could not be ruled out.
To assess whether or not the fetal kidney and ureter findings were linked to parental association, a variety of factors were considered. Macroscopic examination of all adults (22 adults each at 0, 250, 500, and 1000 mg/kg/day) showed that all had paired kidneys and ureters; fetal findings were not associated with similar maternal observations in the affected fetuses. There was also no incidence of absent kidney and/or ureter in any fetus at 250 mg/kg/day. The parental records of the males that sired the affected litters and the family relationship of the does to the stock bucks at 500 and 1000 mg/kg/day were investigated to determine a genetic cause of the cluster of abnormalities in these litters. The investigation showed that the males and females were unrelated (10 generations) and there was no common link to the paternal males in the affected fetuses.
In addition, there were no other major abnormalities (malformations) in the three affected fetuses, no evidence of maternal toxicity, and AGIQ did not adversely affect the rate of resorption, post-implantation loss, mean number of live pups per litter, mean fetal weight, or total number of malformed fetuses. Furthermore, unilateral kidney agenesis often co-occurs with malformation of the ipsilateral uterus or uterine tube in females and epididymis, vas deferens, or seminal vesicles in males.35 No such abnormalities were observed in the three affected fetuses. Most organ systems, including the urogenital systems of both sexes, develop with bilateral symmetry. There are plausible mechanisms that could explain bilateral kidney agenesis, such as interference with synthesis or delivery of glial-cell-derived neurotrophic factor at the time of induction of the ureteric bud at approximately GD 11.5, but not the unilateral condition seen in this study.37 While all of the affected fetuses also presented with skeletal variations, nearly all fetuses in all treated and control groups also exhibited skeletal variations and no other malformations or anomalies were present. Thus, the collective data from Phase I suggested that AGIQ did not exert a typical teratogenic response on the maternal-fetal unit (i.e., a continuum of effects spanning offspring death, structural malformations, and growth retardation). Therefore, the findings of unilateral kidney agenesis in the absence of other expected effects are considered equivocal and not likely due to AGIQ treatment.
To further assess whether or not the higher incidence of absent fetal kidney and ureter at 500 and 1000 mg/kg/day in Phase I was related to treatment, a second phase (Phase II) focused on examination of kidneys and ureter was conducted at those dose levels only, incorporating larger groups (48 animals each in the control, 500, and 1000 mg/kg/day groups) to increase the observational power of the investigation.
In Phase II, lower mean maternal food intake was 77% of control at 1000 mg/kg/day, possibly related to test agent palatability, and there was yellow-orange staining of the cage tray paper and the fur of treated animals. Overall mean maternal body weight gain (GD 6–29) at 1000 mg/kg/day was statistically significantly lower than control, presumably related to decreased food intake. The macroscopic examination in this phase was limited to the presence, absence, and/or potential abnormality of paired kidneys and ureters in the adults and fetuses. The investigation revealed all adults (144) had paired kidneys and ureters and all fetuses (1028; comprising 349, 317, and 362 in the control, 500, and 1000 mg/kg/day groups, respectively) had paired kidneys and ureters, with the exception of one control fetus that had a single kidney and ureter. This follow-up investigation showed that the marginally higher incidence of unilateral kidney/ureter agenesis observed in Phase I at 500 and 1000 mg/kg/day was not a reproducible treatment-related event. In addition, the occurrence of unilateral kidney/ureter agenesis in the control group during Phase II (along with the findings in Phase I) may indicate a potential shift in genetic predisposition to this finding in this supply of rabbits. Therefore, given the lack of reproducibility for kidney/ureter agenesis in Phase II under a more robust sample evaluation (>2 × the sample size per group) and the presence of kidney/ureter agenesis in the control group of Phase II, the renal and ureter observations noted in Phase I are attributed to chance and considered unrelated to treatment with AGIQ.
Minor and non-adverse skeletal fetal abnormalities were evident at 1000 mg/kg/day (Phase I) and consisted of an increase in the incidence of full supernumerary 13th rib, 20 thoracolumbar vertebrae, unilateral caudal shift of the pelvic girdle and a slight increase in the incidence of incompletely ossified cervical vertebral elements and epiphyses. The reduced fetal skeletal ossification at 1000 mg/kg/day correlated with the slightly lower mean fetal weight noted at this dose level and, while potentially treatment-related, is considered non-adverse because it is minor in nature and considered transient. Rib and vertebral findings are also minor and common findings without apparent adverse consequences.
When considering the collective findings from Phase I and Phase II, oral gavage administration of AGIQ to pregnant NZW rabbits at dose levels of 250, 500, and 1000 mg/kg/day was well-tolerated. There was no adverse effect of treatment on the outcome of pregnancy or embryo-fetal survival, growth, or development; therefore, the no-observed-adverse-effect-level (NOAEL) for maternal toxicity and embryo-fetal survival, growth, and development is 1000 mg/kg/day.
Acknowledgements
This work was conducted at Covance Laboratories Limited. Covance was responsible for the study design, the collection, analysis, and interpretation of data, and preparation of the final study reports. Teratology consultation was provided by W Breslin, Breslin Toxicology Consulting, LLC and D Donahue, Integrated Laboratory Services (ILS), Inc. The draft manuscript was prepared by L Kaufman, a paid consultant of ILS. The authors acknowledge the contributions of staff members at Covance who provided technical, animal care, formulation, dosing, necropsy, analytical chemistry, and quality assurance services in support of this work.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AGIQ is a product of San-Ei Gen F.F.I., Inc. (SEG). S Hayashi is a scientific advisor to SEG. R Maronpot is a paid consultant of SEG. Study management was arranged by Integrated Laboratory Systems, Inc. (ILS) and W Breslin and D Donahue were paid by ILS. A Leggett is an employee of Covance Laboratories Limited, which was contracted to perform the study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by San-Ei Gen F.F.I., Inc., Osaka, Japan.
ORCID iD
Robert R Maronpot  https://orcid.org/0000-0003-0653-3484
https://orcid.org/0000-0003-0653-3484
References
1. Amado, NG, Cerqueira, DM, Menezes, FS, et al. Isoquercitrin isolated from Hyptis fasciculata reduces glioblastoma cell proliferation and changes beta-catenin cellular localization. Anticancer Drugs 2009; 20: 543–552.
2. Junior, AG, Gasparotto, FM, Lourenço, ELB, et al. Antihypertensive effects of isoquercitrin and extracts from Tropaeolum majus L: evidence for the inhibition of angiotensin converting enzyme. J Ethnopharmacol 2011; 134: 363–372.
3. Kim, Y, Narayanan, S, Chang, K-O. Inhibition of influenza virus replication by plant-derived isoquercetin. Antiviral Res 2010; 88: 227–235.
4. Li, R, Yuan, C, Dong, C, et al. In vivo antioxidative effect of isoquercitrin on cadmium-induced oxidative damage to mouse liver and kidney. Naunyn Schmiedebergs Arch Pharmacol 2011; 383: 437–445.
5. Nyska, A, Hayashi, S-M, Koyanagi, M, et al. Ninety-day toxicity and single-dose toxicokinetics study of alpha-glycosyl isoquercitrin in Sprague-Dawley rats. Food Chem Toxicol 2016; 97: 354–366.
6. Valentová, K, Vrba, J, Bancířová, M, et al. Isoquercitrin: pharmacology, toxicology, and metabolism. Food Chem Toxicol 2014; 68: 267–282.
7. Hobbs, CA, Koyanagi, M, Swartz, C, et al. Comprehensive evaluation of the flavonol anti-oxidants, alpha-glycosyl isoquercitrin and isoquercitrin, for genotoxic potential. Food Chem Toxicol 2018; 113: 218–227.
8. Erlund, I, Kosonen, T, Alfthan, G, et al. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. Eur J Clin Pharmacol 2000; 56: 545–553.
9. Manach, C, Morand, C, Demigné, C, et al. Bioavailability of rutin and quercetin in rats. FEBS Lett 1997; 409: 12–16.
10. Hasumura, M, Yasuhara, K, Tamura, T, et al. Evaluation of the toxicity of enzymatically decomposed rutin with 13-weeks dietary administration to Wistar rats. Food Chem Toxicol 2004; 42: 439–444.
11. Ishikura, Y, Fujii, W, Sakakibara, Y, et al. Safety evaluation of excessive intake of the drink containing Japanese pagoda tree polyphenol (enzymatically modified isoquercitrin) in healthy adults include obesity persons. Jpn Pharmacol Ther 2008; 36: 931–939.
12. Salim, EI, Kaneko, M, Wanibuchi, H, et al. Lack of carcinogenicity of enzymatically modified isoquercitrin in F344/DuCrj rats. Food Chem Toxicol 2004; 42: 1949–1969.
13. Tamano, S, Hatahara, Y, Sano, M, et al. 13-week oral toxicity and 4-week recovery study of enzymatically modified isoquercitrin in F344/DuCrj rats. Jpn J Food Chem Saf 2002; 8: 161–167.
14. Maronpot, RR, Ramot, Y, Koyanagi, M, et al. Ten-day and four-week toxicity and toxicokinetics studies of alpha-glycosyl isoquercitrin in juvenile Göttingen minipigs. Toxicol Res Appl 2019; 3: 2397847319855087.
15. OECD . Test No. 414: Prenatal developmental toxicity study. Paris: OECD Publishing, 2018.
16. U.S. EPA . Health Effects Test Guidelines: OPPTS 870.3700 Prenatal Developmental Toxicity Study [EPA 712–C–98–207]. United States Environmental Protection Agency (U.S. EPA), 1998.
17. Japanese MAFF . Test data for registration of agricultural chemicals, 12 Nohsan No. 8147. Japanese Ministry of Agriculture, Forestry and Fisheries (MAFF) Agricultural Production Bureau, 2000.
18. Good Laboratory Practice Regulations 1999 . UK Statutory Instrument 1999 No. 3106, https://www.legislation.gov.uk/uksi/1999/3106/contents/made (1999, accessed 12 May 2020).
19. Good Laboratory Practice (Codification Amendments Etc.) Regulations 2004 . UK Statutory Instrument 2004 No. 994, https://www.legislation.gov.uk/uksi/2004/994/contents/made (2004, accessed 12 May 2020).
20. OECD . OECD principles on good laboratory practice (as revised in 1997), ENV/MC/CHEM(98)17. Paris, France: Organisation for Economic Co-operation and Development (OECD), 1998.
21. Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances (codified version) (Text with EEA relevance) . Document 02004L0010-20090420, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02004L0010-20090420 (2004, accessed 12 May 2020).
22. Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 . UK Statutory Instrument 2012 No. 3039, https://www.legislation.gov.uk/uksi/2012/3039/contents/made (2012, accessed 12 May 2020).
23. Bartlett, MS . Properties of sufficiency and statistical tests. Proc R Soc Lond Ser Math Phys Sci 1937; 160: 268–282.
24. Williams, DA . A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 1971; 27: 103–117.
25. Williams, DA . The comparison of several dose levels with a zero dose control. Biometrics 1972; 28: 519–531.
26. Dunnett, CW . A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc 1955; 50: 1096–1121.
27. Dunnett, CW . New tables for multiple comparisons with a control. Biometrics 1964; 20: 482–491.
28. Kruskal, WH, Wallis, WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc 1952; 47: 583–621.
29. Kruskal, WH, Wallis, WA. Errata for Kruskal–Wallis. J Am Stat Assoc 1953; 48: 907–911.
30. Wilcoxon, F . Individual comparisons by ranking methods. Biom Bull 1945; 1: 80–83.
31. Shirley, E . A non-parametric equivalent of Williams’ test for contrasting increasing dose levels of a treatment. Biometrics 1977; 33: 386–389.
32. Steel, RGD . A multiple comparison rank sum test: treatments versus control. Biometrics 1959; 15: 560–572.
33. Fisher, RA . Statistical methods for research workers. 14th ed. New York, NY: Hafner Publishing Company, 1973.
34. Lipsitz, SR, Laird, NM, Harrington, DP. Generalized estimating equations for correlated binary data: using the odds ratio as a measure of association. Biometrika 1991; 78: 153–160.
35. Shapiro, E, Goldfarb, DA, Ritchey, ML. The congenital and acquired solitary kidney. Rev Urol 2003; 5: 2–8.
36. Davis, JP, Koyanagi, M, Maronpot, RR, et al. Identification of compound causing yellow bone discoloration following alpha-glycosyl isoquercitrin exposure in Sprague-Dawley rats. Arch Toxicol 2000; 94: 2413–2421.
37. DeSesso, JM. Comparative gestational milestones in vertebrate development.. In: Hood, RD ed Developmental and reproductive toxicology: a practical approach. 3rd ed. London: Informa Healthcare, 2012, pp. 93–138 [Chapter 6].