
Rodents have been used extensively in virtually all fields of biomedical research and have been the primary species used in toxicologic and carcinogenic research. Over many years it has become obvious that some conditions and in particular some tumors in rodents have questionable relevance in humans. Some of these include peroxisome proliferatoractivator receptor-α (PPAR-α) agonist-induced liver tumors, alpha2μ-globulin-induced renal tumors in male rats, and bladder tumors induced in rats by urinary calculi. In this chapter we review the human relevance of Leydig cell tumors (LCTs), which have been induced in rodents by a number of compounds. We will consider the similarities and differences between humans and rats in the physiology of the Leydig cell (LC) and the pathology of LCTs. Most importantly, we will examine the mechanisms of action that induce LCTs in rats and humans and present data on incidence, physiology, human endocrine disease, and comparative epidemiological studies that strongly indicate LCTs in rodents, in particular the rat, are of little relevance to human health.
Development
The ontogeny of the LC can be reviewed in two basic ways: by following the stages of adult LC differentiation or by examining the two recognized generations of LCs, fetal and adult. Differentiation of the LC is typically broken down into four stages: stem LCs, progenitor LCs, immature LCs, and adult LCs. Stem LCs are spindle shaped and as a cell, which has not yet committed to a lineage of development, it does not express the LC-specific markers such as luteinizing hormone receptor or steroidogenic enzymes such as 3β-hydroxysteroid dehydrogenase. In the rat, at postnatal day 14 stem LCs begin to stain positive for 3β-hydroxysteroid dehydrogenase and are identified at this point as progenitor LCs. These cells develop from about postnatal day 14 until day 28 and begin to produce androgen. (Reviewed in Chen et al., 2009) Gene expression changes most when stem LCs develop into progenitor LCs, whereas differences in gene expression from progenitor to immature LCs and from immature to adult LCs are minimal, suggesting these cells are relatively more similar (Stanley et al., 2011).
Starting at postnatal day 28, progenitor LCs transform morphologically to become more round. The immature LC has increased amounts of smooth endoplasmic reticulum and steroidogenic enzyme levels. Testosterone is not yet the major product produced by these cells because they possess high levels of androgen-metabolizing enzymes that produce 5α-androstane-3α, 17β-diol. The immature LC population doubles once from postnatal day 28 to 56, at which point they develop into adult LCs. The androgen-metabolizing enzyme activity reduces while synthesis of testosterone increases. By day 90, testosterone production in an adult LC of a rat is 150 times than that of a progenitor, and five times than that of an immature LC (Shan et al., 1993).
Development of LCs can also be discussed in reference to the two generally recognized generations: fetal and adult. In the rat, the fetal LC begins to produce testosterone at about gestation day 16 and peaks on day 19 (Habert and Picon, 1984). In humans, testosterone peaks at the end of the 1st trimester (Reyes et al., 1974). The production of androgens is critical for masculinization of the fetus during what is termed the “masculinization programming window.” In the rat this window is from 15.5 to 17.5 days of gestation while in the human it extends from 8 to 14 weeks of gestation. (Welsh et al., 2008) In addition to androgens, fetal LCs produce insulin-like growth factor 3, which is responsible for testicular descent (Zimmerman et al., 1999).
A number of factors that influence the differentiation of the fetal LCs have recently been identified and include: desert hedgehog, a cell signaling molecule secreted by Sertoli cells, and transcription factors such as GATA-4. Interestingly, in rodents, development of fetal LCs is not dependent on pituitary luteinizing hormone (LH) (until the hypothalamic–pituitary– gonadal axis begins functioning near birth). Primates, including humans, have a brief period of independence from hormonal stimulation but then fetal LCs become dependent on placental chorionic gonadotropins (Reviewed in O’Shaughnessy and Fowler, 2011; Svechnikov et al., 2010).
The adult LC forms mostly during puberty and produces the testosterone responsible for spermatogenesis, along with differentiation of other secondary sex characteristics. In both the rat and human there is decreased testosterone production associated with aging. In humans the decreased testosterone is associated with increased levels of LH and appears to be due to a decline in the number of LCs through degeneration. In rats, however, decreased testosterone is associated with declining LH levels and seems to be related to the inability of the LC to respond to LH stimulation (Reviewed in Chen et al., 2009; Cook et al., 1999).
Steroidogenesis
The LC is responsible for producing virtually all of the steroids of the testis; testosterone being the major steroid (Stocco, 1996). Although a number of pathways have been elucidated recently, the primary signaling method for steroidogenesis is initiated when LH binds to a G proteincoupled receptor, which in turn activates adenylate cyclase, producing cAMP (See Figure 109.1). Protein kinase A is activated by cAMP and results in phosphorylation of a number of proteins and transcription factors (Wang and Ascoli, 1990). One of the most important of these is steroidogenic acute regulatory protein (StAR), which is estimated to mediate 85–90% of steroid synthesis (Reviewed in Stocco et al., 2005). Steroidogenic acute regulatory protein and translocator protein, formerly known as peripheral-type benzodiazepine receptor, are components of a large, multiprotein complex known as the transducesome. This complex is responsible for transporting cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane (Reviewed in Rone et al., 2009). The translocation of cholesterol from the outer to inner mitochondrial membrane is the rate limiting step in the synthesis of all steroids (Miller, 2007; Manna 2009). Intracellular cholesterol is produced by three mechanisms: de novo synthesis in the endoplasmic reticulum; mobilization from the plasma membrane and uptake from circulating cholesterol esters; and mobilization from lipid droplets (Rone et al., 2009).
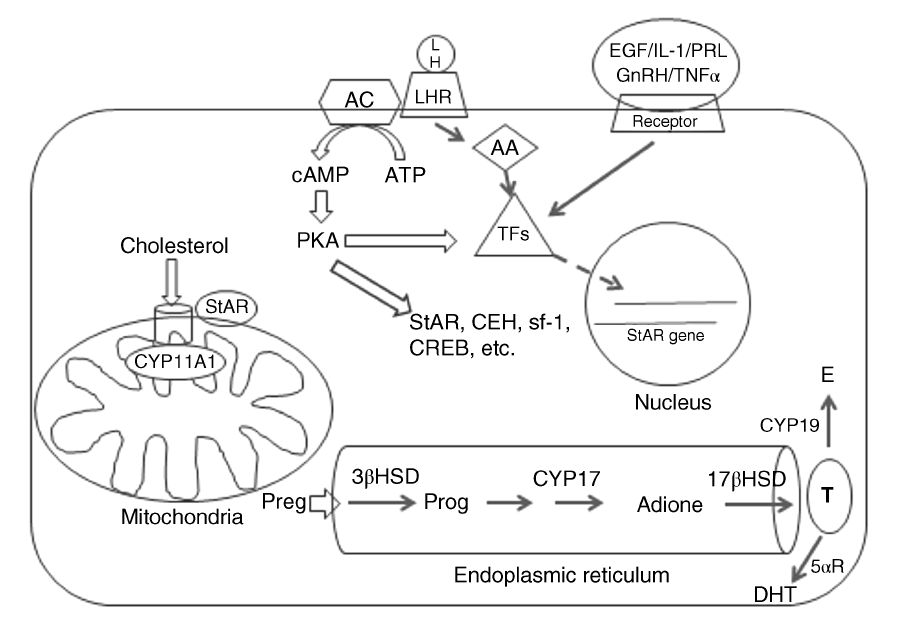
FIGURE 109.1 Mechanisms involved in the synthesis of testosterone.
Mediated by StAR, free cholesterol is transported to the inner mitochondrial membrane, where it is metabolized into pregnenolone by the P450 cholesterol side-chain cleavage enzyme (CYP11A1) (Reviewed in Payne and Hales, 2004). The remaining reactions of steroidogenesis occur within the endoplasmic reticulum with small differences between rats and humans in the pathway (Cook et al., 1999). In rat pregnenolone is transformed to progesterone by the enzyme 3β-hydroxysteroid dehydrogenase (3βHSD). Next the P450 enzyme, C17a-hydroxylase/C17-20-lysase (CYP17), catalyzes two reactions converting progesterone first to hydroxyprogesterone and then to C19 steroid, androstenedione. In humans, CYP17 prefers pregnenolone as a substrate and converts it to 17α-hydroxypregnenolone and then to C19 steroid, dehydroepiandrosterone (DHEA). Finally, 17β-hydroxysteroid dehydrogenase (17βHSD) converts the C19 steroid into testosterone (Payne and Hales, 2004). Testosterone can be further metabolized into estrogen by P450 aromatase (CYP19) (Payne and Hales, 2004) or into the more potent androgen dihydrotestosterone (DHT) (Cook et al., 1999).
As mentioned, cAMP-dependent activation of protein kinase A is essential for activation of StAR and other proteins and transcription factors involved in steroidogenesis, to include cholesterol esterase (CEH), steroidogenic factor-1 (sf-1), GATA-4, and cAMP response element-binding protein (CREB). However, there are other mechanisms by which steroidogenesis is regulated. In recent years research has focused on factors that influence and regulate StAR, since it is critical to the rate-limiting step of steroidogenesis. A number of factors can regulate StAR via cAMP-independent pathways. These include epidermal growth factor (EGF), macrophage-derived factors such as IL-1 and TNFα, hormones such as prolactin and gonadotropin-releasing hormone (GnRH), chloride ions, and calcium messenger systems. In addition, a protein kinase C pathway can be activated resulting in increased transcription and translation of StAR (Stocco et al., 2005; Manna et al., 2007). Leutinizing hormone binding to its receptor can release arachidonic acid (AA) and metabolites of AA can regulate StAR gene expression (Wang et al., 1999).
Pathology
The mammalian testis is composed of a fibrous tunic that surrounds convoluted seminiferous tubules and supporting stroma. In a microscopic section the tubules make up the majority of the cellular structure present and contain mostly gametes in various stages of differentiation from spermatagonia to spermatozoa in addition to supportive cells, such as Sertoli cells. Surrounding the tubules is a small amount of connective tissue that contains blood vessels, nerves, and interstitial cells of Leydig (Figure 109.2). Normal LCs have a wide variation in morphology. They can be spindle shaped with little cytoplasm but are typically round and contain large amounts of lightly eosinophilic cytoplasm. In humans and the wild bush rat, the LC cytoplasm contains elongated, cigar-shaped structures known as crystals of Reinke. These crystals can be seen with light microscopy and are found only in adults. Their origin and function are unknown (Young and Heath, 2000).
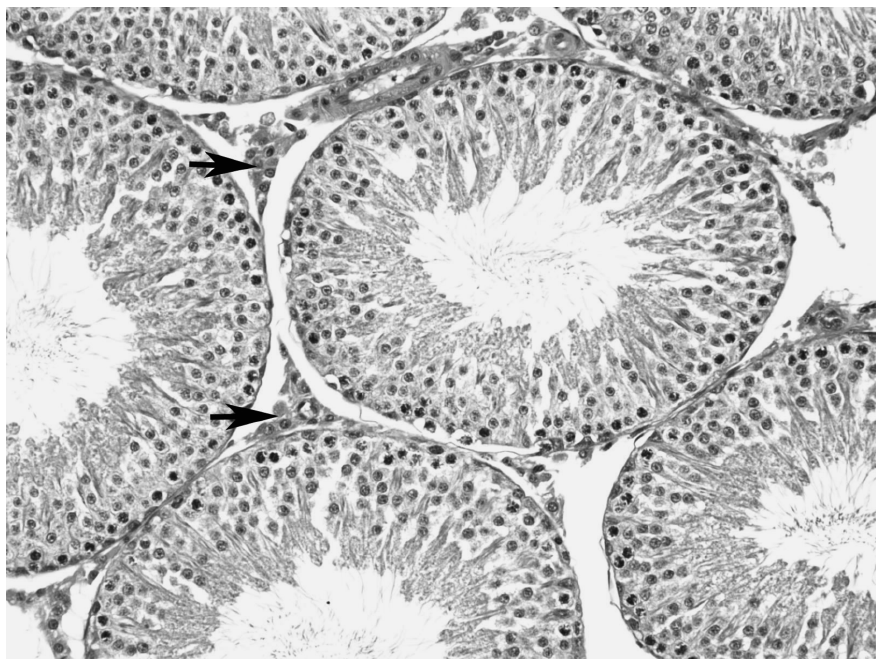
FIGURE 109.2 There are low numbers of Leydig cells in the interstitium surrounding seminiferous tubules of this normal rat testis (H&E, 200×).
In rodents, hyperplasia of LCs is characterized by aggregates of cells that focally or diffusely expand the interstitium between seminiferous tubules. Cells are typically round with centrally located nuclei and granular, eosinophilic cytoplasm. Occasionally, spindle cells with little cytoplasm and dark nuclei are also found (Boorman et al., 1990).
Leydig cell adenoma begins as hyperplasia and consists of one or more foci of expansile LCs that typically compress adjacent tubules. The distinction between hyperplasia and adenoma is difficult as cellular characteristics cannot distinguish between them. Most often size is used as a morphologic criteria to differentiate hyperplasia from adenoma. One recommendation from the National Toxicologic Program (NTP) is to use the size of adjacent tubules as a threshold. When the lesion grows larger than the diameter of the adjacent tubule, it is diagnosed as adenoma. In 2005 toxicologic pathology societies from around the world began an initiative to standardize nomenclature for proliferative and nonproliferative lesions in rats and mice. This initiative is termed the International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice (INHAND) (Mann et al., 2012). The INHAND focuses on specific organ systems. For the male reproductive system, the INHAND guidance differentiates hyperplasia from adenoma using a size of three seminiferous tubules, among other criteria (Creasy et al., 2012). This makes interpretation of incidence data in rodents difficult as published data on LC adenoma typically does not describe the methodology used in establishing the diagnosis of adenoma (Cook et al., 1999).
The incidence of LC adenoma in rodents is highly dependent on species and strain and increases with age. In mice it is typically low, <2.5%, whereas in Sprague-Dawley and Wistar rats it ranges from 4 to 7% (Cook et al., 1999). In Fisher 344 rats, however, the incidence of LC adenoma approaches 100% if the rat is allowed to live its life span (Boorman et al., 1990). LC tumors in rodents are almost always benign as malignancy is rarely reported (Boorman et al., 1990; Cook et al., 1999).
In contrast to rodents, LC tumors in humans are rare, comprising from 1 to 3% of all testicular neoplasms with an estimated incidence of 0.1–3 per million (Cook et al., 1999). Comparing the incidence of LCTs between rodents and humans is somewhat problematic. The data in rodents comes from toxicologic and carcinogenic studies, where rodents are subjected to complete histologic examination and small tumors, not evident grossly, are often discovered microscopically. In humans, testicular tumors are most often diagnosed by palpation and routine microscopic examination of testicular tissue does not occur. Advanced imaging techniques, however, have made possible the diagnosis of non-palpable testicular tumors. Recent data has shown that when ultrasound is used to diagnose testicular tumors, the incidence of LCT is significantly higher than previously reported (Leonharsberger et al., 2011). Regardless, even when considering detection bias, the incidence of LCT in rodents, especially in some strains, is markedly higher than in humans. Furthermore, LCTs in humans are found in all ages, from prepubertal boys to older men. Microscopically they resemble tumors in rodents except for the presence or Reinke crystals. While the majority of human LCTs are benign, a much higher incidence, 10%, are considered malignant in humans. (Cook et al., 1999)
Mechanism Of Tumor Induction
In the rat there are a number of plausible mechanisms for the induction of LC tumors. These include agonists of estrogen, GnRH, dopamine receptors, and PPAR-α. In addition, androgen receptor antagonists, and inhibitors of testosterone synthesis and the enzymes aromatase and 5α-reductase can lead to LCTs. Most of these mechanisms are related to elevated levels of LH. It is well documented that prolonged elevated levels of LH induce LC hyperplasia and neoplasia in the rat (Neumann, 1991; Prentice and Meikle, 1995; Huseby, 1996). In addition, administration of testosterone to F344 rats lowers LH levels and blocks LCT formation (Chatani et al., 1990).
Because LH plays such a central role in LCTs of rats, it is useful to review the hypothalamic-pituitary-testis (HPT) axis to see how LH levels can influence the induction of LCTs. Figure 109.3, (adapted from Cook et al., 1999), highlights five mechanisms by which chemicals can disrupt LH levels and induce LCTs. These five mechanisms, and others, will be briefly reviewed in more detail and additional compounds known to induce LCTs in rats will be listed.
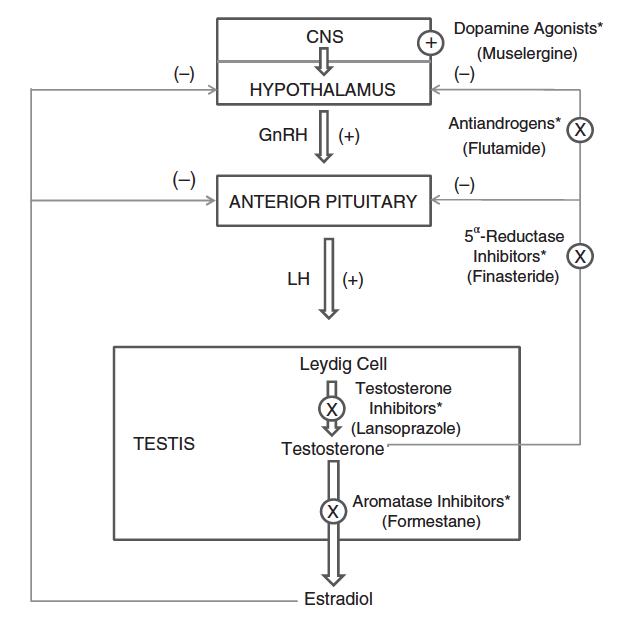
FIGURE 109.3 Regulation of the HPT axis and control points for potential disruption.
Flutamide is an androgen receptor antagonist. It competes with testosterone and DHT for binding at the androgen receptor (Simard et al., 1986). As a result, the androgen signal to the hypothalamus and pituitary is reduced, which stimulates an increase in LH secretion to counter the decreased androgen levels (Cook et al., 1993; ViguierMartinez et al., 1983). Examples of other androgen receptor antagonists known to induce LCTs in rats include cimetidine (Leslie et al., 1981); procymidone (Murakami et al., 1995); and bicalutamide (Iswaran et al., 1997).
In a similar manner, 5α-reductase inhibitors, such as finasteride block the conversion of testosterone to DHT (Prahalada et al., 1994; Rittmaster et al., 1992). Dihydrotestosterone has a higher binding affinity to the androgen receptor than does testosterone (DeGroot et al., 1995). Thus a decrease in DHT reduces the net androgenic signal to the hypothalamus and pituitary resulting in a compensatory increase in LH. Interestingly, 5α-reductase inhibitors induce LCTs in mice and LC hyperplasia in rats (Cook et al., 1999).
Inhibition of testosterone synthesis also lowers the androgen signal resulting in increased LH levels. Interestingly, ketoconazole, which is the most widely known inhibitor of testosterone synthesis, did not induce LCTs but this seemingly was due to the design of the study, which exposed animals to lower levels and for shorter durations than similar bioassays (Cook et al., 1999). Examples of compounds that induce LCTs by inhibiting testosterone synthesis include lansoprazole (Fort et al., 1995); calcium channel blockers such metronidazole (Rustia and Shubik, 1979).
Formestane is an aromatase inhibitor and as such it blocks the conversion of testosterone to estradiol. Since estradiol provides negative feedback to the hypothalamus and pituitary (see Figure 109.3), inhibition of estradiol, in theory, would increase the levels of LH and induce LCTs. However, formestane induced LC hyperplasia in beagle dogs but not rodents (Junker-Walker and Nogues, 1994) and aminoglutethimide, the most well-known aromatase inhibitor, did not induce LC hyperplasia or LCTs (Salhanick, 1982; Shaw et al., 1988). The role of aromatase inhibitors in leading to LCTs, therefore, is equivocal.
Dopamine agonists such as muselergine decrease prolactin levels, which causes downregulation of LH receptors on LCs (Prentice et al., 1992). The decrease in LH receptors lowers the overall testosterone production and LH levels increase to compensate. An alternative mechanism was proposed based on work with oxolinic acid. Yamanda et al. suggested that dopamine agonists increase levels of GnRH and thereby increase LH levels (Yamanda et al., 1995). The exact mechanism has yet to be worked out.
In rats GnRH induces LCTs at low doses but not at higher doses. This is because the LC of the rat contains GnRH receptors and can stimulate the LC directly. Low levels of GnRH can also increase LH through direct stimulation of the pituitary (Figure 109.3). At higher doses, negative feedback inhibition from GnRH would lower LH levels and thus LCTs. (Cook et al., 1999) Mice and humans do not have GnRH receptors on their LCs and are thus not susceptible to LCTs by GnRH agonists (Hunter et al., 1982).
Estrogen agonists induce LCTs in the mouse but almost never in the rat (Cook et al., 1999). However, PPAR-α agonists are known to lead to LCTs in rats. In at least some of the cases of PPAR-α agonists there is an increase in estradiol (but not LH or testosterone) that corresponds with the potency of the compound (Biegel et al., 2001). PPAR-α agonists also can reduce testosterone levels (Cook et al., 1992; Gazouli et al., 2002). As shown previously, compounds that lower testosterone can cause a compensatory rise in LH and lead to LCTs. Most likely, both elevation of estrogen and decreased testosterone play a role in induction of LCTs by PPAR-α agonists (Klaunig et al., 2003).
Human Relevance
It is clear from the forgoing discussion that many compounds induce LCT in rodents, especially rats. Since the rat is one of the primary species used in toxicologic and carcinogenic risk assessment, these findings may be of importance in establishing exposure thresholds or compound safety data. However, a number of factors, when examined closely, illustrate that the occurrence of LCTs in rats is not biologically relevant to humans. These differences are thoroughly discussed and referenced in Cook et al. (1999) and the reader is referred there for a more in depth analysis. This discussion will focus on differences in comparative biology, tumor incidence, as well as cases of human genetic disease and data from epidemiology studies.
The biology of the rat and human differ in a number of important ways and some of those potentially have an impact on the relevance of LCTs in humans. These include differences in serum proteins, response to hCG, number of LH and GnRH receptors, sensitivity to prolactin, and LH-related aging changes.
The sex hormone-binding globulin is absent in the rat. It is produced in the liver in humans and binds to the majority of testosterone. This binding lowers the metabolism and clearance of testosterone in humans and dampens short-term changes in testosterone levels. In contrast, in the rat testosterone levels can be more rapidly altered and as a result, compounds that alter testosterone levels have a more dramatic effect in rats.
Rats and humans have been shown to respond differently to hCG, a hormone that has equivalent effects on LCs as does LH. In vivo and in vitro data show that hCG induces LC hyperplasia in rats while inducing hypertrophy in humans. In addition, testosterone secretion and the mitogenic response are up to 100-fold less in human LCs.
There are important differences between rats and humans in the receptors on LCs. Compared to humans, rats have over 10 times the number of LH receptors on their LCs. This spare capacity of receptors allows for a lower concentration of LH in rats that will induce a response in LCs. In addition, as mentioned, rats have GnRH receptors on LCs, while humans do not. It would appear, therefore, that induction of LCTs by GnRH agonists is not a mechanism applicable to humans. Likewise, unlike rats, humans do not express prolactin receptors on LCs. In the rat, dopamine agonists act by decreasing prolactin levels, which in turn lower the number of LH receptors on their LCs. The decrease in LH receptors results in a decrease in the testosterone levels and a compensatory increase in LH. Because humans lack prolactin receptors, dopamine agonists seemingly would not alter the number of LH receptors on human LCs and testosterone, and LH levels would remain unchanged. Therefore, it is unlikely that dopamine agonists would induce LCTs in humans.
Finally, rats and humans differ in how LH affects testosterone levels in aging. In both rats and humans testosterone levels decrease over time. However, in rats LH levels also decrease and this decrease in LH may be responsible for the decrease in testosterone. In contrast, LH levels remain unchanged in men with aging. Luteinizing hormone appears, therefore, not to be a factor in decreasing the level of testosterone in men with time.
As previously discussed, the incidence of LCT in rats and humans is markedly different. Depending on the strain, the incidence of LCT in rats ranges from nearly 100% in Fisher 344s to 5% in Wistar (Tucker, 1997) and 6.5% for SpragueDawley (McMartin et al., 1992) rats. In contrast, in humans testicular cancer accounts for about 1% of all cancers in men and of these, only about 1% are of LC origin; making LCTs about 0.01% of all cancers in men (Cook et al., 1999). Consequently, the incidence of LCT in humans is in the order of 1.9 million-fold less than in F344 and 130,000-fold less than Sprague-Dawley rats (Cook et al., 1999).
There are two genetic endocrine diseases in humans that highlight the mechanistic differences in LCT induction between humans and rats. The first is androgen insensitivity syndrome, which is due to a defect in the androgen receptor. The second, familial male precocious puberty, is the result of a defect in the LH receptor. Reviewing the incidence of LCTs in individuals affected with these diseases further emphasizes that humans are clearly less sensitive to LCT formation than are rats.
In androgen insensitivity syndrome there is a genetic defect in the androgen receptor so that androgens are not recognized. The syndrome ranges from complete to partial insensitivity and phenotypically, affected individuals range from infertile males to females. Individuals with complete insensitivity and a cryptorchid testis have elevated levels of LH, but normal levels of testosterone. Testicular tumors, including LCTs, are more common in individuals with androgen insensitivity syndrome with some reports of LC adenomas as high as 2–3%. As a comparison, the incidence of LCT in rats when administered an androgen receptor antagonist, such as flutamide, is almost 100%. Administration of flutamide to rats can be thought of as a chemically induced form of androgen insensitivity syndrome. If humans were as sensitive to the formation of LCTs, then individuals with this syndrome would have a much higher incidence of LCTs, which they clearly do not. (Cook et al., 1999)
Familial male precocious puberty is the result of a genetic mutation of the LH receptor that results in constitutive activation. Affected individuals undergo puberty at about 4 years of age. They have testosterone production and LC hyperplasia but undetectable levels of LH. The continuous activation of LH receptors in individuals with familial male precocious puberty mimics the primary mechanism of LCT induction in rats, namely elevated LH levels. While LC hyperplasia is reported in cases of familial male precocious puberty, there have been no documented cases of LCTs despite a lifetime of LH-receptor activation. Once again, if humans were as sensitive to LH-induced LCT formation, individuals with familial male precocious puberty would have a much higher incidence of LCT (Cook et al., 1999).
Lastly, there have been a number of epidemiological studies conducted on compounds with human exposure for which there is comparative rodent study data available. Only three will be reviewed to illustrate the concept but for a more complete list refer to Cook et al. (1999). It must be emphasized that the epidemiological surveys relied on insensitive methods for the detection of LCT in humans but nonetheless the results are still helpful in underscoring the relative difference in LCT incidence between humans and rodents.
1,3-Butadiene is a ubiquitous product used in the plastics industry and because of its high exposure to humans, it has been extensively studied both in laboratory animals and humans. Sprague-Dawley rats exposed via inhalation had an increased incidence of LCTs (8% vs. 0% in controls) (Owen and Glaister, 1990). Using a large number of exposed workers, human epidemiological studies have failed to identify a risk of LCTs in men exposed to 1,3-butadiene (Divine and Hartman, 2001). Cadmium induces not only LCTs in rats but also adenomas in the prostate (Waalkes et al., 1988). However, a review of human exposure studies identifies increased risk of lung cancer but not prostate or LCTs (Verougstraete et al., 2003). Finally, trichloroethylene has wide human exposure as an industrial solvent and an additive in food, drugs, and consumer products. It causes marked increases in LCTs in Sprague-Dawley rats but a review of five cohort studies in humans did not identify trichloroethylene as a carcinogen (Cook et al., 1999).
Conclusion
In rats, LCTs are induced almost exclusively by elevated levels of LH. The exceptions seem to be LCTs induced by GnRH and dopamine agonists, which, as discussed, appear not to be applicable to humans since they lack the receptors (GnRH and prolactin) to induce LCTs. Therefore, when examining the relevance of LCTs induced in rats to human health, the question becomes what is the relative sensitivity in humans to LCT induction from elevated LH levels. As discussed, several lines of reasoning indicate that the high incidence of LCTs in rats is not relevant to humans. First, humans have a markedly lower incidence of LCTs compared to rodents, especially rats. Second, there are a number of comparative differences in the biology of the LC, especially the type and number of receptors that help explain the difference in rates of LCTs. Third, two endocrine diseases in humans do not produce LCTs at an incidence level expected, if the mechanism of LCT induction in human was similar to that in rats. And fourth, when compounds with both human epidemiological and rodent exposure data are compared, the results do not support a conclusion that rodent LCTs are relevant to human health.
References
Biegel, L.G., et al. (2001) Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol. Sci. 60(1):44–55.
Boorman, G.A., et al. (1990) Testis and epididymis. In: Boorman, G.A., et al., editors. Pathology of the Fisher Rat, San Diego, CA: Academic Press.
Chatani, F., et al. (1990) Stimulatory effect of a lutenizing hormone on the development and maintenance of 5α-reduced steroid- producing testicular interstitial cell tumors in Fisher 344 rats. Anticancer Res. 10:337–342.
Chen, H., Ge, R.S., and Zirkin, B.R. (2009) Leydig cells: from stem cells to aging. Mol. Cell. Endocrinol. 306(1–2):9–16.
Cook, J.C., et al. (1992) Induction of Leydig cell adenomas by ammonium perfluorooctanoate: a possible endocrine-related mechanism. Toxicol. Appl. Pharmacol. 113(2):209–217.
Cook, J.C., et al. (1993) Investigation of a mechanism of Leydig cell tumorigenesis by linuron in rats. Toxicol. Appl. Pharmacol. 119:195–204.
Cook, J.C., et al. (1999) Rodent Leydig cell tumorigenesis: a review of the physiology, pathology, mechanisms, and relevance to humans. Crit. Rev. Toxicol. 29(2):169–261.
Creasy, D., et al. (2012) Proliferative and nonproliferative lesions of the rat and mouse male reproductive system. Toxicol. Pathol. 40:40S–121S.
DeGroot, L.J., et al. (1995) Endocrinology, 3rd ed., W.B. Saunders, Philadelphia.
Divine, B.J. and Hartman, C.M. (2001) A cohort mortality study among workers at a 1,3 butadiene facility. Chem. Biol. Interact. 135–136:535–553.
Fort, F.L., et al. (1995) Mechanism for species-specific induction of Leydig cell tumors in rats by lansoprazole. Fundam. Appl. Toxicol. 26(2):191–202.
Gazouli, M., et al. (2002) Effect of peroxisome proliferators on Leydig cell peripheral-type benzodiazepine receptor gene expression, hormone-stimulated cholesterol transport, and steroidogenesis: role of the peroxisome proliferator-activator receptor alpha. Endocrinology 143(7):2571–2583.
Habert, F. and Picon, R. (1984) Testosterone, dihydrotestosterone and estradiol-17 beta levels in maternal and fetal plasma and in fetal testes in the rat. J. Steroid Biochem. 21(2):193–198.
Hunter, M.G., et al. (1982) Stimulation and inhibition by LHRH analogues of cultured rat Leydig cell function and lack of effect on mouse Leydig cells. Mol. Cell Endocrinol. 27:31–34.
Huseby, R.A. (1996) Leydig cell neoplasia. In: Payne, A.H., Hardy, M.P., and Russel, L.D., editors. The Leydig Cell, Vienna, IL: Cache River Press.
Iswaran, T.J., et al. (1997) An overview of animal toxicology studies with bicalutamide (ICI 176,334). J. Toxicol. Sci. 22(2):75–88.
Junker Walker, U. and Nogues V. (1994) Changes induced by treatment with aromatase inhibitors in testicular Leydig cells of rats and dogs. Exp. Toxic. Pathol. 46:211–213.
Klaunig, J.E., et al. (2003) PPAR-α agonist-induced rodent tumors: modes of action and human relevance. Crit. Rev. Toxicol. 33(6):655–780.
Leonharsberger, L., et al. (2011) Increased incidence of Leydig cell tumors of the testis in the era of improved imaging techniques. BJU Int. 108:1603–1607.
Leslie G.B., et al. (1981) A two-year study with cimetidine in the rat: assessment for chronic toxicity and carcinogenecity. Toxicol. Appl. Pharmacol. 61:119–137.
Mann, P.C., et al. (2012) International harmonization of toxicologic pathology nomenclature: an overview and review of basic principles. Toxicol. Pathol. 40:7S–13S.
Manna, P.R., Dyson, M.T., and Stocco, D.M. (2009) Regulation of the steroidogenic acute regulatory protein gene expression: pres- ent and future perspectives. Mol. Hum. Reprod. 15(6):321–333.
Manna, P.R., Jo, Y., and Stocco, D.M. (2007) Regulation of Leydig cell steroidogenesis by extracellular signal-regulated kinase 1/2: role of protein kinase A and protein kinase C signaling. J. Endocrinol. 193(1):53–63.
McMartin, D.N., et al. (1992) Neoplasms and related proliferative lesions in control Sprague-Dawley rats from carcinogenicity studies. Historical data and diagnostic considerations. Toxicol. Pathol. 20(2):212–225.
Miller, W.L. (2007) Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim. Biophys. Acta 1771(6):663–676.
Murakami, M., et al. (1995) Species-specific mechanism in rat Leydig cell tumorigenesis by procymidone. Toxicol. Appl. Pharmacol. 131(2):244–252.
Neumann, F. (1991) Early indicators for carcinogenesis in sex hormone-sensitive organs. Mutat. Res. 248:341–356.
O’Shaughnessy, P.J. and Fowler, P.A. (2011) Endocrinology of the mammalian fetal testis. Reproduction 141:37–46.
Owen, P.E. and Glaister, J.R. (1990) Inhalation toxicity and carci- nogenicity of 1,3-butadiene in Sprague-Dawley rats. Environ. Health Perspect. 86:19–25.
Payne, A.H. and Hales, D.B. (2004) Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25(6):947–970.
Prahalada, S., et al. (1994) Leydig cell hyperplasia and adenomas in mice treated with finastride, a 5 alpha-reductase inhibitor: a possible mechanism. Fundam. Appl. Toxicol. 22(2):211–219.
Prentice, D.D., et al. (1992) Mesulergine induced Leydig cell tumors, a syndrome involving the pituitary-testicular axis of the rat. Arch. Toxicol. 15(Suppl.):197–204.
Prentice, D.E. and Meikle, A.W. (1995) A review of drug-induced Leydig cell hyperplasia and neoplasia in the rat and some comparisons with man. Hum. Exp. Toxicol. 14:562–572.
Reyes, F.L., et al. (1974) Studies on human sexual development. II. Fetal and maternal serum gonadotropin and sex steroid concen- trations. J. Clin. Endocrinol. Metab. 38(4):612–617.
Rittmaster, R.S., et al. (1992) Effect of finastride, a 5 alpha-reductase inhibitor, on serum gonadotropins in normal men. J. Clin. Endo- crinol. Metab. 75(2):484–488.
Roberts S.A., et al. (1989) SDZ 200-100 induces Leydig cell tumors by increasing gonadotrophins in rats. J. Am. Coll. Toxicol. 8:487–505.
Rone, M.B., Fan, J., and Papadopoulos, V. (2009) Cholesterol transport in steroid biosynthesis: role of protein-protein inter- actions and implications in disease states. Biochim. Biophys. Acta 1791(7):646–658.
Rustia, M. and Shubik, P. (1979) Experimental induction of hepa- tomas, mammary tumors, and other tumors with metronidazole in noninbred Sas:MRC(WI)BR rats. JNCI 63:863–868.
Salhanick, H.A. (1982) Basic studies on aminoglutethimide. Cancer Res. 42:3315s–3321s.
Shan, L.X., et al. (1993) Differential regulation of steroidogenic enzymes during differentiation optimizes testosterone produc- tion by adult rat Leydig cells. Endocrinology 133(5):2277–2283.
Shaw, M.A., Nicholls, P.A., and Smith, H.J. (1988) Aminoglute- thimide and ketoconazole: historical perspectives and future prospects. J. Steroid Biochem. 31:137–146.
Simard, J., et al. (1986) Characteristics of interaction of the anti- androgen flutamide with the androgen receptor in various target tissues. Mol. Cell. Endocrinol. 44(3):261–270.
Stanley, E.L., et al. (2011) Stem Leydig cell differentiation: gene expression during development of the adult rat population of Leydig cells. Biol. Reprod. 85:1161–1166.
Stocco, D.M. (1996) Acute regulation of Leydig cell steroido- genesis. In: Payne, A.H., Hardy, M.P., and Russel, L.D., editors. The Leydig Cell, Vienna, IL: Cache River Press.
Stocco, D.M., et al. (2005) Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: more complicated than we thought. Mol. Endocri- nol. 19(11):2647–2659.
Svechnikov, K., et al. (2010) Origin, development and regulation of human Leydig cells. Horm. Res. Paediatr. 73:93–101.
Tucker, M.J. (1997) The male genital system. In: Diseases of the Wistar Rat, Bristol, PA: Taylor & Francis.
Verougstraete, V., et al. (2003) Cadmium, lung and prostate cancer: a systematic review of recent epidemiological data. J. Toxicol. Environ. Health B Crit. Rev. 6(3):227–255.
Viguier-Martinez M.C., et al. (1983) Effect of a non-steroidal antiandrogen, flutamide, on the hypothalmo-pituitary axis, geni- tal tract and testis in growing rats: Endocrinological and histo- logical data. Acta Endocrinologica 102:299–306.
Waalkes, M.P., et al. (1988) Cadmium carcinogenesis in male Wistar
Wang, H.Y. and Ascoli, M. (1990) Reduced gonadotropin responses in a novel clonal strain of Leydig tumor cells estab- lished by transfection of MA-10 cells with a mutant gene of a type I regulatory subunit of the cAMP-dependent protein kinase. Mol. Endocrinol. 4(1):80–90.
Wang, X., Walsh, L.P., and Stocco, D.M. (1999) The role of arachidonic acid on LH-stimulated steroidogenesis and steroido- genic acute regulatory protein accumulation in MA-10 mouse Leydig tumor cells. Endocrine 10(1):7–12.
Welsh, M., et al. (2008) Identification in rats of a programming window for reproductive tract masculinization, disruption of which leads to hypospadias and cryptorchidism. J. Clin. Invest. 118:1479–1490.
Yamada, T. et al. (1995) A possible mechanism for the increase of serum leutinzing hormone levels in rats by oxolinic acid. Toxicol. Appl. Pharmacol. 134:35–42.
Young, B. and Heath, J.W. (2000) Wheater’s Functional Histology, London: Harcourt Publishers.
Zimmerman, S., et al. (1999) Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol. Endocrinol. 13:681–691.