
There is a vast amount of literature related to mouse liver tumorigenesis generated over the past 60 years, not all of which has been captured here. The studies reported in this literature have generally been state of the art at the time they were carried out. A PubMed search on the topic “mouse liver tumors” covering the past 10 years yields over 7000 scientific papers. This review address several important topics related to the unresolved controversy regarding the relevance of mouse liver tumor responses observed in cancer bioassays. The inherent mouse strain differential sensitivities to hepatocarcinogenesis largely parallel the strain susceptibility to chemically induced liver neoplasia. The effects of phenobarbital and halogenated hydrocarbons in mouse hepatocarcinogenesis have been summarized because of recurring interest and numerous publications on these topics. No single simple paradigm fully explains differential mouse strain responses, which can vary more than 50-fold among inbred strains. In addition to inherent genetics, modifying factors including cell cycle balance, enzyme induction, DNA methylation, oncogenes and suppressor genes, diet, and intercellular communication influence susceptibility to spontaneous and induced mouse hepatocarcinogenesis. Comments are offered on the evaluation, interpretation, and relevance of mouse liver tumor responses in the context of cancer bioassays.
Introduction
Since the early 1970s and even before, the mouse liver tumor response observed in cancer bioassays has been a source of unresolved controversy with books, symposia, and advisory committee deliberations on the topic. Some have taken the debate further and argued that the mouse bioassay should be eliminated altogether from our hazard identification/safety assessment armamentarium and that a one species default model, the rat, is sufficient. Debates on a one or two species bioassay and the relevance of the mouse liver tumor response continue.
Our knowledge of factors associated with mouse hepatocarcinogenesis has significantly improved over the last two decades. The diagnostic and nomenclature issues ofconcern in the 1970’s have been largely resolved. The propensity of male mice versus females to develop spontaneous and treatment-induced liver tumors is better understood. Advances in the genetics of strain susceptibility have been made, although more work remains to tease out the definitive genes responsible. Modulators of murine hepatocarcinogenesis, such as diet, hormones, oncogenes, methylation, imprinting, and cell proliferation/apoptosis are among multiple mechanistically associated factors that impact this target organ response in control as well as treated mice. With the advancement of our understanding, it has become obvious that no mode of action, pathway, or mechanism should be considered mutually exclusive.
There is no one simple paradigm to explain the differential strain sensitivity to hepatocarcinogenesis. With the recent effort to delineate over 8 million single nucleotide polymorphisms in 15 mouse strains selected for genetic re-sequencing, the prospect for continued and possibly specialized use of mice for hazard identification and safety assessment remains. In the meantime, we need to continue ongoing efforts and progress in defining threshold limiting factors that impact murine hepatocarcinogenesis and use this knowledge to place the mouse liver tumor response in appropriate regulatory perspective. There will always be a need for research on mode of action and quantitative differences/similarities between species.
Origins of Inbred Mice
Classical strains used in research today were originally derived from interbreeding of Mus musculus subspecies. It first started as a hobby in 18th century Victorian England and Asia with breeding of “fancy” mice based on coat color and the subsequent adoption of some of these mice in the early 1900’s by researchers in the United States. The oldest inbred strain, the DBA/2 (originally dba for d = dilute, b = brown, and a = non-agouti) was established in 1909 by C. C. Little. Ten years later the mouse lady of Granby, Massachusetts, Miss Abbie E. C. Lathrop, intercrossed black offspring of a female 57 mouse to produce a stock of mice ultimately used by Clarence Cook Little to form his C line resulting in the C57BL/6. Strains A, C3H, and CBA were created by Leonell C. Strong in the early 1940’s.
Commonly used mouse strains
In the early 1960’s the National Cancer Institute adopted the B6C3F1 mouse, the F1 hybrid of the C57BL/6 female and C3H male, as the mouse for use in the cancer bioassay program. The decision to use the B6C3F1 mouse was based on results from an 18-month study involving 20,000 mice including two hybrid lines and 127 different chemicals. Partially based on historic inertia and partly based on a concern about losing the historic control database, the B6C3F1 has remained the mouse model of choice in U.S. Government-sponsored hazard identification programs for toxicity and cancer. Variations of the C57BL and the outbred Swiss stock are popular models for safety assessment by the chemical and pharmaceutical industries while these plus the A strain, B6CF1, C3H, BALB/c, FVB, 129, and others are used by biomedical researchers. The recent publication of mouse haplotype maps for 15 inbred mouse strains, including 8 classical strains commonly used for research, is expected to facilitate obtaining important information for a wide range of biological questions and may influence how mice are used for hazard identification in the future.
Experimental Protocols/Models for Mouse Liver Tumor Induction
Conventional hazard identification bioassay
While the hazard identification paradigm of maintaining a core group of 50 animals per dose per sex has remained relatively constant for many years, modifications such as include three to five doses, some of which approximate human exposure levels, in utero exposures and stop studies have been added to answer specific questions. The basic hazard identification study commences exposures when mice and rats are 6 to 8 weeks of age and treatment typically continues for 2 years. When using specifically susceptible mouse strains such as the B6C3F1 hybrid, relatively high and variable incidences of liver tumors can occur in the untreated or vehicle control mice. The liver tumor incidence increases as the mice age with the majority of liver tumors occurring after 20 months of age.
Single or multiple doses to adult mice
Administration of a few to several repeated doses of known and usually genotoxic hepatocarcinogens, starting with 5 to 8-week old mice will yield liver tumors in a few months, making this a potentially useful tool to follow liver tumor pathogenesis and to study variables that might influence hepatocarcinogenesis. Published reports using this type of protocol were fairly common prior to the 1970s with a general switch to initiation-promotion studies in subsequent years. It should be noted, however, that repeated and prolonged exposures to nongenotoxic agents certainly can result in induction of mouse liver tumors.
Neonatal mouse model
While variations of this model have been used, the basic approach is to administer one to three doses of the test agent prior to weaning with no further treatment. The rationale is that “fixation” of hepatocellular initiation will be favored by the high rate of cell growth during the neonatal period. Subsequent endogenous promotion would allow for clonal expansion of any initiated clones with development of liver tumors within a 12-month observation period. This model, using either CD1 or B6C3F1 mice, has been described and shown to work well with genotoxic test agents. A single dose of a hepatocarcinogen such as diethylnitrosamine to a neonate at 12 to 15 days of age will yield a high incidence and multiplicity of liver neoplasia with a relatively short latency while the same dose given to the adult mouse will result in a lower incidence and multiplicity with a much longer latency period. B6C3F1 and C3AF1 mice neonatally treated with ethylnitrosourea versus treatment at 42 days of age results in a 3- to 8-fold increase in liver tumors in the neonates compared to the older dosed mice. This age difference is based upon an efficient initiation of hepatocytes during the neonatal period when there is rapid liver growth. The neonatal mouse model has been used extensively to learn more about the biology of murine hepatocarcinogenesis and has included studies on the genetics of susceptibility, the relative roles of cell proliferation and apoptosis, effects of hormones and diets on liver tumor development, and the effects of DNA methylation on expression of genes relevant to murine hepatocarcinogenesis.
Initiation-promotion protocols
These protocols typically consist of administration of the initiating agent, usually diethylnitrosamine, to either neonatal mice or to mice shortly after weaning followed by repeated dosing with an agent being tested as a liver tumor promoter. One variation involves administration of a necrogenic dose of diethylnitrosamine 1 to 2 weeks after weaning. Another variation involves partial hepatectomy of 5 to 6 week old mice followed in 36 hours by a non-necrogenic dose of diethylnitrosamine. These two protocols without further treatment will generally yield 3 or more liver tumors per mouse at one year of age. A third protocol, the preweaning initiation-promotion protocol, involves administration of a non-necrogenic dose of diethylnitrosamine or an alternative initiator often at age day 12 or 15 followed by administering the agent being tested for promotion shortly after weaning. Without further treatment, these mice can develop a high multiplicity of liver tumors by one year of age while treatment with a promoter significantly shortens the time to tumor. Partial hepatectomy alone after preweaning initiation also serves as a liver tumor promoter. While these initiation-promotion protocols can be carried out until there is an obvious liver tumor endpoint, quantitation of putative preneoplastic foci of cellular alteration has been used as a biomarker of hepatocarcinogenicity. Features of these initiation-promotion protocols have been described in a review paper dealing with phenobarbital promotion. There are variations such as initiation by treatment with diethylnitrosamine in drinking water for 4 weeks followed by promotion and other examples will be referenced throughout this document.
Genetically engineered mouse models
Several lines of viral oncogene induced mouse models of hepatic neoplasia are summarized by Sandgren. These include ras and myc oncogenes under the influence of the albumin gene regulatory elements as well as SV40-TAg driven by a variety of regulatory elements. Growth factors such as TGFalpha and IGF2 under the influence of the metallothionein-1 or major urinary protein regulatory elements, respectively, yield dysplastic foci in as little as 6 months and hepatocellular neoplasms in 8 to 18 months with evidence of severe hepatic dysplasia in newborn mice.
In an effort to more closely mimic human hepatitis-associated hepatocellular neoplasia, a small number of genetically engineered mouse models based on incorporation of various portions of the human hepatitis virus such as the HB × gene and the pre-S2 gene have been developed and show a male predominance under the influence of androgens and glucocorticoids. Hepatic changes can appear as early as 4 weeks and consist of glycogen-rich centrilobular foci of cellular alteration. During a latent period of several months the altered foci show enhanced cell proliferation and aneuploidy with ultimate development of hepatocellular carcinomas adjacent to adenomas and additional foci of cellular alteration. The yield of hepatocellular carcinomas can exceed 80% and an X gene/c-myc construct will yield dysplastic foci in 12 weeks and hepatocellular carcinomas between 20 and 28 weeks. In essentially all of the HBV models of HCC, there is an imbalance of cell proliferation and apoptosis.
B6C3F1 mice expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2 develop hepatopathy and are prone to lymphoid neoplasia as well as heptocellular carcinoma. Lymphoid neoplasia was seen after age 18 months. Hepatopathy was present at 12 months and tumors later (exact age of occurrence of hepatocellular carcinomas not stated).
The genetically engineered mouse models that have been proposed for hazard identification, such as the rasH2 and p53+/-, fall into the category of hepatocarcinogenesis resistant mice.
Strain Susceptibility to Spontaneous and Induced Liver Tumors: Biology
Based on genetic and numerous other factors, mouse stocks and inbred strains differ in susceptibility to both spontaneous and treatment-induced liver neoplasia. Because of the variety of studies with differing protocols used to generate susceptibility data, direct comparisons among strains and stocks is problematic and, in looking across the numerous published studies, the best we can do is to classify mice by their relative susceptibility to hepatocarcinogenesis. By way of example, CBA and C3H inbred mice are considered highly susceptible to induction of liver neoplasia while in comparison C57BL/6 and BALB/c are relatively resistant.
There are two metrics typically used to categorize liver tumor susceptibility: incidence and multiplicity. The former is most often used to characterize spontaneous liver neoplasia and the latter to assign relative susceptibility to treatment-induced liver neoplasia. A study by Hanigan and co-workers serves to demonstrate the magnitude of relative susceptibility. Utilizing an identical protocol, a direct comparison between C3H/HeJ and C57BL/6J male mice with respect to liver tumor induction demonstrated up to a 40-fold difference in liver tumor multiplicity.
In defining resistance and susceptibility (i.e., strain dependent variablilty), two factors play a significant role – the number of initiated tumor cells and the rate of tumor growth. Using the basal levels as an indicator of potential spontaneous initiation in mouse liver, 8-hydroxydeoxyguanosine (OH8dG) strain differences in C3H, B6C3F1, and C57BL mice are positively correlated with spontaneous hepatocellular neoplasia. Since formation of OH8dG has been shown to result in gene mutation and hypomethylation, it may also contribute via these mechanisms to the differential strain susceptibility to spontaneous hepatocarcinogenesis. C3H mice are more susceptible than C57BL/6 mice to a variety of hepatocarcinogens with differing metabolic activation patterns and they form similar numbers of preneoplastic lesions and DNA adducts, indicating that the relative susceptibility of these mice is based on detection of proliferative lesions which in turn is dependent upon lesion growth rate within the temporal context of the experimental study.
Spontaneous liver tumors
The strain-specific spontaneous incidence of liver tumors (originally diagnosed as hepatomas) in mice has been documented in the published literature since the late 1930’s and early 1940’s. Andervont reported on the occurrence of spontaneous hepatomas in C3H mice, with Burns and Schenken publishing in the following year. Subsequent reports of C3H sublines maintained in different laboratories and over time have confirmed the high spontaneous incidence of liver tumors. The high susceptibility of CBA mice was noted in 1936 and has since been confirmed by others.
Data on tumor incidences including liver tumors for other strains and various F1 hybrids have been published (LP, 129, DBA/2, C57BL/10 -; BALB/c, C57BL/6-). These studies confirm the low incidence of spontaneous hepatocellular neoplasia in LP, 129, DBA/2, BALB/c, and C57BL mice.
Attempts at comparative tabulation of the incidences in these reports of spontaneous liver tumor incidence among different mouse strains can be potentially misleading since countless variables including study design, diet, caging, diagnostic features, and study duration differ among these reports.
Treatment induced liver tumors
Strain sensitivity to treatment induced liver tumors generally parallels strain susceptibility to spontaneous liver tumor development. Susceptibility to treatment-induced liver neoplasia is contingent upon the temporal timing and frequency of dosing as well as the magnitude of the dose and the duration of the study observation period. Thus, a single dose of a hepatocarcinogen such as diethylnitrosamine to a neonate at 15 days of age will yield a high incidence and multiplicity of liver neoplasia with a short latency while the same dose given to the adult mouse will result in a lower incidence and multiplicity with a much longer latency period. A 10-fold increase in diethylnitrosamine (5 to 50 mg/kg) resulted in a 3.7-fold increase in number of tumors but with size distribution similar at the low and high dose. Swiss mice initiated neonatally with ethylnitrosourea at two different doses also had a dose-dependent increase in liver tumors as did B6C3F1 mice initiated with dimethylnitrosamine, and BALB/c mice treated with 2-acetylaminofluorene. It was also reported that the higher doses favored the development of malignant over benign liver tumors.
The number of papers dealing with treatment-induced hepatocarcinogenesis and the various treatment regimens are legion. A few illustrative examples to highlight strain susceptibility will be provided.
The sensitivity of C3H mice to chemical induction of liver tumors began to be documented shortly after their susceptibility to spontaneous liver tumors was known. Heston and coworkers showed increased susceptibility to liver tumors in C3H mice treated with urethane and the antifertility drug enovid. In more recent years, studies at the McArdle lab by Drinkwater and coworkers as well as in the laboratory of Dragani and coworkers in Milan have studied the genetics and modifying factors responsible for differential strain sensitivity to murine hepatocarcinogenesis.
Using a neonatal mouse model in which the mice were injected no later than 16 hours after birth, it was shown that 9, 10-dimethyl-1,2-benzanthracene caused a pronounced increased incidence of liver tumors in male and female C3H, CBA, C3H × CBA, and CBA × C3H but only a marginal increase in strain A, C57BL, A × C57BL, C57BL × A, BALB/c and IF. Liver tumor latency was the shortest in male C3H mice.
Both diethylnitrosamine and ethylnitrosourea have been used in studies of treatment-induced mouse hepatocarcinogenesis. Vesselinovitch published several studies in the 1970s primarily using the B6C3F1 preweaning initiation-promotion mouse model with ethylnitrosourea. Lee used this model to study diethylnitrosamine induced liver tumors in C3H–C57BL/6 chimeras. A comparison of 11 inbred strains using ethylnitrosourea and/or diethylnitrosamine was reported by the McArdle lab.
While treated males typically are more susceptible to liver tumor induction than treated females, occasional exceptions do occur. Male and female C3H mice treated with carbon tetrachloride by gavage and 4-o-tolylazo-o-toluidine by subcutaneous injection develop increased incidences of hepatomas with the female response to 4-o-tolylazo-o-toluidine exceeding that of the treated males. Another example of a prominent female response is seen In diethylnitrosamine treated C57BR/cdJ mice.
DBA/2 susceptibility
Male DBA/2 mice represent an unusual and puzzling situation with respect to liver tumor susceptibility. The spontaneous liver tumor rate is a low 1.5% and treatment of 5-week-old male DBA/2 mice with diethylnitrosamine yields a relatively low liver tumor response. However, treatment of 12-day old DBA/2 mice with diethylnitrosamine results in a 20-fold increase in liver tumor multiplicity versus liver tumor multiplicity in the resistant C57BL/6 mouse. The reason for this differential in susceptibility is not known but differences in metabolism have been speculated as playing a role.
Two specific categories of treatment induced murine liver tumors, viz., halogenated hydrocarbons and phenobarbital, are highlighted separately because of general academic and regulatory interest.
Halogenated hydrocarbons
Halogenated hydrocarbons continue to receive scientific attention because of potential widespread human exposure to contaminated drinking water as well as to water disinfection by-products. This class of compounds is clearly associated with liver tumor induction in mice and typically there is no clear evidence of carcinogenicity in other species, including rats. There are multiple modes of action believed to be contributory to the murine hepatocarcinogenicity associated with one or more of the halogenated hydrocarbons. These include enhanced cell proliferation, decreased apoptosis, perturbation of DNA methylation, disruption of gap junctional intercellular communication, activation of oncogenes, and peroxisome proliferation.
Often used as a classic example of the enhanced cell proliferation secondary to a cytotoxicity mode of action, chloroform has been classified as possibly carcinogenic to humans (IARC Classification 2B). While corn oil gavage of chloroform leads to murine liver tumors in B6C3F1 male and female mice, chloroform given in drinking water is not hepatocarcinogenic. This differences is attributed to differential hepatotoxicity and increased hepatocellular proliferation in gavage studies. Similarly, when chloroform was given by drinking water in an initiation-promotion study, it inhibited liver tumor development compared with a positive liver tumor response with phenobarbital promotion. Cytotoxicity and secondary enhanced cell proliferation has been proposed as a primary mode of action for three other trihalomethanes (bromodichloromethane, chlorodibromomethane, and bromoform).
The mode of action of the murine hepatocarcinogen trichloroethylene is extensively reviewed by Bull. Trichloroethylene causes liver tumors in B6C3F1 and Swiss mice. The metabolism of trichloroethylene to chloral hydrate, dichloroacetic acid, and trichloroacetic acid complicates teasing out the modes of action since these metabolites are also hepatocarcinogenic in mice. Modification of cell signaling pathways that alter cell replication and cell death as a consequence of decreased DNA methylation as well as c-mycand c-jun methylation are likely contributory modes of action for the tumorigenic effects of trichloroethylene. Studies of the trichloroethylene metabolites dichloroacetic acid, trichloroacetic acid, and chloral hydrate suggest that both dichloroacetic acid and trichloroacetic acid are involved in trichloroethylene-induced liver tumorigenesis and that many dichloroacetic acid effects are consistent with conditions that increase the risk of liver cancer in humans. Some of these effects involve GST Xi, histone methylation, and overexpression of IGF2.
While there are differences in the type of dose response for dichloroacetic acid and trichloroacetic acid induced mouse liver tumors, both dichloroacetic acid and trichloroacetic acid have caused liver tumors in multiple mouse studies. Several studies have examined the modes of action involved in dichloroacetic acid induced murine hepatocarcinogenesis and have implicated hypomethylation, enhanced cell proliferation, peroxisome proliferation or even activation of PPAR without evidence of peroxisome proliferation, and oncogene activation. Based upon hypomethylation identified in mouse liver tumors following initiation with methylnitrosourea and promotion by dichloroacetic acid or trichloroacetic acid, and as an early response in short term exposure to dichloroacetic acid and trichloroacetic acid, hypomethylation is clearly involved in murine hepatocarcinogenesis associated with these chemicals and can be blocked by dietary methionine.
A comprehensive review of aldrin/dieldrin has been published. Dieldrin is considered a ground water contaminant and, like the water disinfection by-products discussed above, it is associated with mouse liver tumor responses in different mouse strains. Oxidative stress generated via futile cycling of the cytochromes enzymes has been implicated as a primary mode of action responsible for the murine liver tumor response. It has been proposed that the consequences of oxidative stress are threshold responses and should be used in risk assessment for dieldrin and for other halogenated hydrocarbons.
In addition to dieldrin, the organochlorine class of chemicals has been clearly associated with induction of murine liver tumors. Thirty-seven of 138 organochlorine agrochemicals were hepatocarcinogenic in mice without an apparent effect of mouse strain or study duration. A clear positive association between hepatomegaly at one year and a liver tumor response at 18 or 24 months was documented in these studies.
Phenobarbital
Phenobarbital is one of the most widely studied rodent hepatocarcinogens, is considered the prototype chemical for a mode of action whereby hepatic enzyme induction leads to rat and mouse liver tumor induction, and is typically used as a positive control in rodent initiation-promotion protocols. An extensive review of the relationship between phenobarbital and mouse liver neoplasia has been published by McClain. McClain provides information on a series of studies utilizing different phenobarbital treatment protocols from the early 1970s to the early 1980s to test for liver tumor susceptibility in C3H, CF1, B6C3F1, and C57BL mice. As more studies have been carried out from the early 1970s to the present time, it has become obvious that many different factors and variables contribute significantly to phenobarbital induced liver tumors. For example, depending upon the study protocol, phenobarbital may enhance or inhibit liver tumor formation following diethylnitrosamine initiation. Diethylnitrosamine initiation in the postweaning period versus diethylnitrosamine initiation in the preweaning period results in enhancement or inhibition of phenobarbital induced liver tumors, respectively. This paradoxical effect has been carefully studied. The different effect on promotion may be a reflection of the observation that phenobarbital enhances the growth of eosinophilic but not basophilic foci of cellular alteration and proportionally fewer eosinophilic foci are produced with preweaning diethylnitrosamine initiation.
Most studies of the factors involved in phenobarbital associated murine hepatocarcinogenesis have been carried out utilizing initiation-promotion protocols. These studies include examination of differential strain sensitivity, influence of phenobarbital on cell proliferation, oncogene analysis in liver tumors, and effects of phenobarbital on global DNA methylation as well as on methylation of oncogenes. Most of these studies have utilized the preweaning neonatal model of initiation followed by different promotion regimens. The response in these studies is dependent on the strain of mouse used and on the initiating chemical carcinogen. C3H initiated with diethylnitrosamine and given phenobarbital demonstrated an increase in adenomas compared to diethylnitrosamine treatment alone while B6C3F1 males had a decrease in adenomas as a result of phenobarbital treatment. Phenobarbital had no effect on hepatocellular adenomas in C57BL/6 males previously treated with diethylnitrosamine. Following 9, 10-dimethyl-1,2-benzanthracene initiation, phenobarbital increased adenomas in both C3H and B6C3F1 but not in C57BL mice.
In BALB/c mice phenobarbital treatment following initiation by diethylnitrosamine resulted in a decrease latency for heptic adenomas and an increased incidence of adenomas at multiple sampling times.
In comparing C57BL/6, C3H/HeN, and DBA/2 mice initiated with diethylnitrosamine and treated for 17 weeks with phenobarbital, Diwan and coworkers identified an increase in preneoplastic foci and liver tumors in C3H and DBA/2 mice but not in C57BL mice and noted that the DBA/ 2 mice were especially responsive. In a different study using reciprocal C57BL/6 and DBA/2 hybrids, susceptibility to phenobarbital induced tumors was a dominant trait with both F1 hybrids responding similarly.
Timing of phenobarbital dosing appears to be important in how mice respond to diethylnitrosamine initiation and subsequent promotion by phenobarbital. Diethylnitrosamine alone induced focal hepatic lesions, adenomas and carcinomas in B6C3F1 mice but subsequent treatment with phenobarbital suppressed the effect of the diethylnitrosamine, possibly due to some feminizing effect of perinatal administration of phenobarbital. Diethylnitrosamine initiation at 6 and 10 weeks of age versus initiation at 15 days of age with both followed by long-term treatment with phenobarbital resulted in strong liver tumor promotion following initiation at 6 and 10 weeks of age and inhibition of liver tumor development following neonatal initiation in B6C3F1 males. On the other hand, neonatal initiation followed by phenobarbital promotion in BALB/c males enhanced development of liver tumors. Similar effects on inhibition of tumor development by phenobarbital have been seen in B6C3F1 initiated with diethylnitrosamine followed by phenobarbital promotion commencing at 4 weeks of age.
There is considerable evidence to support the differential strain susceptibility to phenobarbital induced murine hepatocarcinogenesis. Diethylnitrosamine injection followed by partial hepatectomy and subsequent dietary phenobarbital resulted in accelerated growth of preneoplastic focal lesions in C3H and BALB/c mice with only slight increased growth of preneoplastic lesions in C57BL/6 mice.
Alterations in the balance between cell proliferation and cell death as they impact phenobarbital hepatocarcinogenesis are influenced by mouse strain. Maximum induction of hepatic DNA synthesis in the absence of any evidence of cytotoxicity in phenobarbital treated 8-week-old B6C3F1 mice is seen in short-term to 28-day studies. This is suggestive of a mitogenic effect. Furthermore, a dose-dependent enhanced DNA synthesis and an associated decreased apoptosis is seen in preneoplastic foci in B6C3F1 mice treated with 100 and 500 mg phenobarbital/kg in the diet but not 10 mg phenobarbital/kg. In an initiation-promotion study in C3H and B3B6F1, cell proliferation was measured in preneoplastic foci and non-involved hepatocytes and there was a differentially enhanced response to phenobarbital in the C3H strain preneoplastic foci. The bromodeoxyuridine labeling index in male C57BL/6J, B6C3F1 and C3H/HeJ mice initiated neonatally with diethylnitrosamine followed by phenobarbital promotion for 12 months was positively correlated with focus growth showing a C3H > C6C3F1 > C56BL/6 strain dependent effect.
Murine strain susceptibility to phenobarbital induced heptocarcinogenesis may also involve alterations in global DNA methylation as well as methylation associated changes in oncogene expression. Both hypomethylation and hypermethylation can be associated with tumorigenesis. While a choline-devoid, methionine-deficient diet causes global DNA hypomethylation in B6C3F1 and in C57BL mice, treatment with phenobarbital lowers DNA methylation in B6C3F1 mice to 20% of control. Phenobarbital treated C57BL mice, on the other hand, maintain normal levels of DNA methylation despite having a higher rate of treatment induced cell proliferation versus B6C3F1. Phenobarbital treatment of B6C3F1 and the two parental strains is primarily associated with hypermethylation changes in GC-rich regions of DNA in the B6C3F1 and C3H mice as an indication that inability to maintain normal methylation is involved with susceptibility to phenobarbital induced liver tumors.
Furthermore, B6C3F1 mice on a choline-devoid, methionine-deficient diet with and without phenobarbital treatment exhibit increased mRNA expression of Ha-ras and raf as a consequence of hypomethylation. There was altered global DNA methylation, both hypomethylation and hypermethylation, in some spontaneous and phenobarbital induced liver tumors as well as increased Ha-ras expression indicative of a decreased ability of the B6C3F1 mouse to maintain its methylation status. Following phenobarbital treatment B6C3F1 mice are also less capable of maintaining methylation of raf in hepatocytes compared to C57BL mice. However, more phenobarbital induced than spontaneous liver tumors had increased raf mRNA levels in contrast to equivalent frequency of enhanced Ha-ras mRNA levels in both phenobarbital and spontaneous tumors indicating that at least for raf, B6C31 mouse liver tumors may arise by a separate pathway from spontaneous tumors.
Examination of the methylation status of Ha-ras, Ki-ras and myc in spontaneous, chloroform and phenobarbital induced liver tumors in B6C3F1 mice showed that Ha-ras was hypomethylated in all tumors examined, Ki-ras was hypomethylated in some tumors, and the methylation status of myc was not changed indicating that there are some common biochemical pathways to spontaneous and induced liver tumorigenesis.
The oncogene mutational profile seen in phenobarbital induced tumors in both C3H and CF1 mice does not differ from the oncogene mutational profile in sponataneous tumors supporting the contention that phenobarbital provides a selective growth advantage to heptocytes with spontaneously occurring mutations.
Very little has been published regarding the hepatocarcinogenicity of phenobarbital in genetically engineered mice. In an initiation-promotion study utilizing 3 different doses of phenobarbital, there was no evidence of hepatic carcinogenicity in a 26-week study usingrasH2 mice on a BALB/c × C57BL/6 F1 background. A 9-month study of phenobarbital in DNA repair deficient XPA -/- mice did not yield any tumors. There were also no liver tumors in XPA -/- × p53 +/- and C57BL/6 mice similarly treated with phenobarbital in the same study. On the other hand, phenobarbital decreased tumor latency and increased multiplicity in livers of c-myc/TGF-alpha mice, primarily as a consequence of phenobarbital blocking cell death during initial stages of tumor development.
Genetics of Murine Liver Tumor Susceptiblity
Although specific murine genes responsible for liver tumor susceptibility have not been discretely identified, several loci that influence susceptibility and resistance to liver tumor induction have been mapped by different research groups using backcrosses and linkage analysis. These loci influence hepatocyte growth control, especially in preneoplastic lesions. The loci and associated mouse chromosomes implicated in mouse hepatocarcinogenesis are presented in Table 1.

Susceptiblity to liver tumor induction can vary more than 50-fold among inbred strains. Quantitative comparison among strain is extremely problematic because of significant differences in animal models used, study protocols, choice of carcinogen, age at dosing, and duration of the observation period. Attempts to tabulate the liver tumor incidences and multiplicities along with identification of all the intended and unintended experimental variables from 70 years of publications would be non-trivial and probably not of much help in fostering an understanding of implications of this target tissue response for human risk assessment. Even qualitative statements of relative sensitivity and susceptibility across decades of studies need to be carefully considered. Most scientists will agree that the C3H male is highly susceptible to liver tumor induction while the C57BL/6 male is highly resistant. Based upon publications of the spontaneous incidences of liver tumors, the C3H male is intrinsically sensitive to develop liver tumors as a function of age while liver tumors are extremely rare in aged C57BL/6 males. Classifying the remaining mouse strains with respect to liver tumor susceptibility is more judgmental but I have attempted to do that in Table 2.

Up to 85% of the greater susceptibility of the C3H versus the C57BL/6 male mouse to liver tumor induction is attributable to an Hcs7 (hepatocarcinogenicity sensitivity 7) locus. Following a neonatal dose of N-ethyl-N-nitrosourea or diethylnitrosamine there was a 1.7- to 2-fold acceleration of the growth rate of preneoplastic foci of cellular alteration in C3H versus C57BL/6 males without further treatment. The Hcs7 locus is also associated with a 2.6-fold higher growth rate of normal hepatocytes in C3H versus C57BL/6 males.
Partial hepatectomy of 6-week old C3H and C57BL/6 mice treated neonatally with N-ethyl-N-nitrosourea results in an accelerated growth rate of preneoplastic lesions in the resistant C57BL/6 mouse but no increase in the already accelerated growth rate of preneoplastic foci in the C3H mice supporting the contention that the Hcs7 locus has a role in hepatocyte growth control. Furthermore, liver tumor multiplicity was greater than 5-fold increased in C57BL/6 males that underwent partial hepatectomy compared to the sham controls while there was a 60% reduction in liver tumor multiplicity in the C3H males that had partial hepatectomy. Thus, partial hepatectomy acted as a promoter for C57BL/6 males but not for C3H males. Partial hepatectomy had no effect on foci or tumors in female mice of either strain. The data suggest that the physiological growth stimulus provided by partial hepatectomy as well as the Hcs7 gene both work through the same growth regulatory pathway.
It has been demonstrated that both the frequency of DNA adducts and number of preneoplastic foci of cellular alteration are similar in C3H and C57BL/6 mice following treatment with N-ethyl-N-nitrosourea and that the hepatocellular foci take longer to grow to easily detectable size in C57BL/6 mice, further supporting the likely role of the Hcs7 locus in preneoplastic lesion growth control. The Hcs7 locus appears to exert its effect at the level of the hepatocyte since diethylnitrosamine-induced liver tumors arise exclusively from C3H heptocytes in C3H-C57BL/6 chimeric mice.
Linkage studies of crosses between C3H and C57BL/6 have shown that the Hcs7 C3H allele is sufficient to render the C57BL/6 susceptible to liver tumor induction with up to a 14-fold increase in liver tumor multiplicity in congenic males. Furthermore, the tumorigenic effect of the Hcs7 C3H allele is independent of gender, causing an increase in tumor multiplicity in congenic females as well as males.
The C57BR/cdJ mouse was originally generated from the same breeding pair that produced the C57BL/6 mouse and paradoxically has a 20-fold greater susceptibility to liver tumor induction. Study of the C57BR/cdJ mouse and related chimeras has led to identification of several loci in addition to Hcs7 that are implicated in hepatocarcinogenesis and account for the enhanced liver tumor susceptiblity of the C57BR/cdJ versus the closely related C57BL/6 mouse.
Female C57BR/cdJ are insensitive to inhibitory effects of estrogen and are unusually susceptible to both spontaneous and treatment-induced liver neoplasia.
The high susceptibility is attributable to 2 loci (Hcf1 and Hcf2; located on chromosomes 17 and 1, respectively). Using C57BR/cdJ–C57BL/6 chimeras, the increased susceptibility was found to be intrinsic to the C57BR/cdJ hepatocytes with over 90% of the tumors in both male and female originating from these hepatocytes. This finding provides evidence that the determinants of hepatocarcinogenesis sensitivity are intrinsic to the specific hepatocytes. Further evidence that the determinants are at the level of the hepatocyte is provided by the early work of Condamine and co-workers using C3H–C57BL/6 and C3H–BALB/c chimeras to examine the cellular composition of liver tumors in aged mice as well as by Lee and co-workers in C3H–C57BL/6 chimeras treated neonatally with diethylnitrosamine.
Multiple genetic loci affecting hepatocarcinogenesis susceptility and resistance have been identified indicating that the genetics underlying susceptibility is complex. Using volume percent as a quantitative index of susceptibility in a study of C3H crosses with A/J and with M. spretus, Dragani and coworkers concluded that strain variation in susceptibility to hepatocarcinogenesis involves polygenic inheritance of unlinked genetic loci.
While basic strain differences in hepatocarcinogen sensitivity are determined by intrinsic genetic factors, studies of C3H–BALB/c sexually chimeric mice treated neonatally with diethylnitrosamine show that male specific hormonal or micro-environmental factors are responsible for promotion of liver cancer in both XX and XY hepatocytes.
Using recombinant inbred, backcross, and intercross mouse breeding schemes, researchers at the McArdle laboratory sought to tease out the genetics responsible for the biological complexity of hepatocarcinogenicity in DBA/2 mice. In the process they identified two hepatocarcinogenesis resistance genes (Hcr1 and Hcr2) in the very sensitive neonatally treated DBA/2 mouse. Based on their linkage analysis which covered ~95% of the genome, it was concluded that neonatally sensitivity DBA/2 mice probably carry multiple hepatocarcinogen sensitive loci, each with a small effect that in the aggregate overcome the resistance conferred by Hcr1 and Hcr2.
In most studies examining the susceptibility of different strains the primary effect is a differential cell proliferative response in putative preneoplastic altered foci during the promotional operational phase of hepatocarcinogenesis. However, in an initiation-promotion study using diethylnitrosamine initiation after partial hepatectomy of males at 6 weeks of age followed by phenobarbital, clofibrate, or ethynyl estradiol promotion, Lee and co-workers found that interstrain differences in both initiation and promotion exist. BALB/c and C57BL/6 had fewer foci of altered hepatocytes than C3H after diethylnitrosamine alone. Phenobarbital accelerated the growth rate of altered foci in both C3H and BALB/c and clofibrate increased the growth of altered foci only in the C3H males.
Mouse strain differential susceptibilities differ between liver and lung tumors following treatment with agents that induce neoplasia in both target sites indicating that genetic factors responsible for liver and lung neoplasia are tissue-specific.
Strain Susceptibility to Spontaneous and Induced Liver Tumors: Modifying Factors
Cell proliferation, apoptosis, and growth kinetics
Cancer development requires an hereditable alteration of DNA plus cellular proliferation. It has been experimentally shown that cell proliferation is a fundamental requirement for initiation, promotion, and progression of spontaneous and treatment induced liver cancer in mice. The basis of the preweaning initiation-promotion model of murine hepatocarcinogenesis is dependent upon agent induced promutagenic DNA damage being “fixed” by the enhanced hepatocellular cell proliferation associated with the rapid liver growth in the neonatal mouse. For the adult mouse, the regenerative hepatocellular proliferative response to hepatonecrogenic doses of chloroform and other nongenotoxic agents, or regenerative growth following partial hepatectomy all serve to initiate the cancer process. It has also been postulated that natural infidelity in DNA maintenance methylation could lead to heritable hypomethylation that would play a functional role in liver tumor initiation. Following initiation, liver cancer promotion requires a proliferative growth advantage for expansion of initiated clones of hepatocytes. During the process of clonal expansion further genetic alterations may yield subclones that have enhanced cell proliferative rates and the ability to progress to malignancy.
There has been considerable study of the relative roles of cell proliferation versus cell death (apopotosis) in murine hepatocarcinogenesis, especially for nongenotoxic liver tumor promoters. It is generally accepted that the critical factor driving growth or regression of preneoplastic hepatocellular lesions in rodents is the balance between cell proliferation and apoptosis both in the preneoplastic lesions as well as in the surrounding noninvolved hepatic parenchyma. In a comparison of the roles of apoptosis and cell proliferation in C3H/He and C57BL/6J mice, it has been reported that cell proliferation is the prevailing determinant of liver tumor promotion by phenobarbital and nafenopin in both strains following diethylnitrosamine initiation.
While enhanced cell proliferation is not a universal predictor of liver carcinogenesis, there is little doubt that it is an important and necessary component of murine hepatocarcinogenesis observed for a spectrum of nongenotoxic agents. Hepatocarcinogenicity may be driven either by enhanced cell proliferation and/or reduced apoptosis within proliferative lesions. The liver tumor response in male B6C3F1 mice attributed to dichloroacetic acid was suggested to involve the ability of dichloroacetic acid to suppress apoptosis rather than to enhance proliferation of initiated cells. Similarly, suppression of apoptosis rather than cell proliferation was attributed to account for the growth of H-ras positive C3H mouse liver tumors. On the other hand, dieldrin promoted focus growth appears dependent upon cell proliferation and without effects on apoptosis at multiple concentrations. In general, murine susceptiblity to peroxisome proliferators as well as other nongenotoxic hepatocarcinogenic agents correlates more strongly with induction of DNA synthesis and less so with suppression of apoptosis.
There is often a concerted effect between cell proliferation and cell death involving preneoplastic foci of cellular alteration and noninvolved hepatocytes and this concerted effect may be influenced by the continuation of treatment. For example, in male B6C3F1 mice dieldrin and phenobarbital increased heptocyte labeling indices and decreased apoptosis in eosinophilic and basophilic lesions and both decreased upon cessation of treatment. Furthermore, the dose of test agent, for example phenobarbital, can influence the relative rates of DNA synthesis and apoptosis with higher doses promoting outgrowth of focal lesions by increased cell proliferation and decreased apoptosis. In an initiation-promotion study using C57BL/6, C6C3F1, and C3H males, phenobarbital promotion following neonatal diethylnitrosamine initiation resulted in a strain-dependent (C3H>B6C3F1>C57BL) increase in focus number and size with focus growth rates positively correlated with cell proliferation and with intrafocal apoptosis occurring late. The authors also suggested that extrafocal apoptosis was contributory to clonal growth via removal of adjacent normal cells.
It is generally accepted that larger proliferative hepatocellular lesions grow faster than smaller lesions. In female B6C3F1 mice initiated with diethylnitrosamine at 12 days of age and subsequently promoted with unleaded gasoline vapor or dietary ethinyl estradiol, larger preneoplastic foci in the treated mice had higher hepatocyte labeling indices. In a hepatocarcinogenesis study involving diethylnitrosamine initiation and phenobarbital promotion of C3H and C3B6F1 mice, hepatocellular adenomas had higher bromodeoxyuridine labeling indices than altered foci with the lowest labeling indices in non-involved hepatocytes. It is noteworthy that the ratio of labeling index in foci to non-involved hepatocytes rather than the level of cell proliferation alone was related to the enhanced liver tumor susceptibility of the C3H mice versus the C3B6F1 hybrid.
Preweaning initiation with ethylnitrosourea followed by a partial hepatectomy as a growth stimulus in C57BL/6J and C3H/HeJ male mice lead to a tumor volume doubling time of 2.2 and 2.9 weeks, respectively. Following neonatal initiation with diethylnitrosamine, three strains of mice were euthanized at 4 intervals up to 42 weeks of age. The number of liver tumors in C3H mice was 2.5 times that in B6C3F1 and C57BL/6 but the growth rates were similar for the 3 strains with a doubling time of 2.1 to 2.5 weeks. Larger tumors grew faster and the tumor periphery grew faster than the central portions, probably due to a gradient of oxygen, nutrition, and growth factors. There were also differences in tumor growth rates among multiple tumors in the same mouse. The growth rate in C57BL/6 liver tumors was initially fast but subsequently slowed down by 80%. It has been suggested that impaired growth of some liver tumors in C57BL/6 mice is associated with accumulation of secretory protein cytoplasmic inclusions in tumor cells.
Different factors may affect hepatoproliferative rates in various studies. Using female B6C3F1 mice, the route of administration can determine whether chloroform enhances cell proliferation in the liver of B6C3F1 mice. Chloroform given by gavage but not in the drinking water increased cell proliferation of hepatocytes. Similarly, the gavage administration of trihalomethanes, including chloroform, bromodichloromethane, chlorodibromomethane, and bromoform, enhanced cell proliferation consistent with their known hepatocarcinogenicity. Dietary restriction of 12-month old male B6C3F1 mice significantly increased the rate of hepatocyte apoptosis and significantly decreased the frequency of proliferating cells compared to ad libitum fed mice.
Enzyme induction
Hepatic enzyme induction has been proposed as a mode of action to explain what is considered a rodent-specific liver tumor response to treatment with phenobarbital and other nongenotoxic rodent hepatocarcinogens. In addition to the publication by McClain, this epigenetic mechanism of hepatocarcinogenesis has been discussed in a 2000 review. Generation of reactive oxygen species as a consequence of futile cycling of P450s is capable of producing tissue necrosis and mutations that would favor development of hepatic neoplasia. Reactive oxygen species can lead to lipid peroxidative damage to hepatocyte cell membranes and may then cause functional alterations in membrane receptors that in turn exert liver promoting action. Reactive oxygen species can also modulate gene expression and lead to altered regulation of growth factors that favor tumor promotion and progression. Temporal increases in hepatic malondialdehyde, a biomarker of oxidative damage to lipids, following dieldrin treatment suggest that oxidative damage may be an early event in dieldrin-induced mouse hepatocarcinogenesis. It has been proposed that proliferative mouse liver lesions induced secondary to enzyme induction, such as eosinophilic nodules seen in phenobarbital treated mice, are phenotypically different from liver tumors seen in control mice or mice treated with genotoxic agents and should not be considered a carcinogenic response directly relevant to the chemical that caused the enzyme induction.
DNA methylation and imprinting
In recent years there has been a wider acknowledgement of the importance and contributory nature of DNA methylation throughout different stages of cancer development. Both global and gene specific alterations involving hypomethylation and hypermethylation of DNA are significant contributory factors to oncogenesis. DNA methylation is required for maintaining the status of imprinted genes and for epigenetic activation of oncogenes and silencing of tumor suppressor genes. Temporal analyses of methylation status in mice used in various hepatocarcinogenesis protocols, particularly in initiation and promotion studies, provide evidence that altered methylation is not just a consequence of malignant transformation but plays a contributory role in liver tumor genesis. I hasten to point out that DNA methylation is involved in a wide variety of biological processes and, consequently, there is no simple explanation that will cover the diverse possible ways methylation can impact oncogenesis. However, there are definite associations between DNA methylation, genomic imprinting, and mouse hepatocarcinogenesis. Further, DNA methylation is influenced by dietary levels of methionine and choline-devoid, methionine deficient induced hypomethylation is quickly reversed by administration of a methionine adequate diet.
The insulin-like growth factor IGF2 and the mannose 6-phosphate(M6P)/IGF2R receptor genes are known to be imprinted, have a monoallelic expression in mice, and provide a possible explanation for enhanced sensitivity of some mice to liver tumor formation. It is also of some relevance that imprinting and alterations in the M6P/IGF2R gene are associated with human liver tumors and that there is loss of IGF2 imprinting in mouse hepatocellular carcinoma cell lines. Genomic imprinting of murine genes regulated by androgen could also play a role in murine hepatocarcinogenesis. The recent creation of conditional knock-out mice with tissue-specific inactivation of murine M6P/IGF2R should prove useful in future studies.
Oncogenes & tumor suppressor genes
Proto-oncogenes are believed to play an important role in the genesis of neoplasia when genetically altered or expressed at increased levels. In conjunction with several other genes, growth factors, and transcription regulatory proteins, proto-oncogenes play pivotal roles in regulating cell growth, differentiation, and development. Oncogene activation associated with mouse liver tumors is both strain and carcinogen dependent. Mouse liver tumors involving several strains document the common observation of mutations in the H-rasprotooncogene with the frequency of H-ras mutations approximately 10-fold higher in genetically susceptible versus resistant strains. For example, codon 61 point mutations in H-ras in diethylnitrosamine induced liver tumors occur with a frequency greater than 50% in C3H mice, 33% in B6C3F1 mice and 0% in C57BL/6 and BALB/c mice. Greater than 50% of spontaneous B6C3F1 and C3H liver tumors harbor H-ras mutations compared to 7% in BALB/c. Analysis of precancerous foci of cellular alteration for evidence of H-rasmutations has shown that a small proportion of these foci have H-ras mutations indicating that this change may be an early and critical event in murine hepatocarcinogenesis. The frequency of activated H-ras in induced mouse liver tumors is typically higher for genotoxic versus nongenotoxic chemicals. For genotoxic hepatocarcinogens there is an inverse dose-response relationship to H-ras activation suggesting that higher doses may at least partially utilize a non-H-ras pathway for tumorigenesis. The low frequency of H-rasmutations in tetrafluoroethylene induced mouse liver tumors is indicative that some mouse liver tumors produced by nongenotoxic chemicals favor development by a ras-independent pathway. The tumor promoters dieldrin and phenobarbital increase the frequency of c-Ha-ras wild-type, but not of c-Ha-ras mutated focal liver lesions in male C3H/ He mice. It has been shown that the preferential outgrowth of proliferative H-ras positive hepatic lesions is mediated by suppression of apoptosis rather than by accelerated rates of cell proliferation.
Other oncogenes can play a role in murine hepatocarcinogenesis. C-jun, which has important functions in cell proliferation and differentiation, is believed to play a role in hepatogenesis and, by implication, in hepatocarcinogenesis. Increased expression of c-myc as well as H-ras has been reported in hyperplastic nodules and hepatocellular carcinomas in B6C3F1 mice treated with dichloroacetic acid and trichloroacetic acid and increased expression of c-junand c-myc in B6C3F1 mice treated with dichloroacetic acid, trichloroacetic acid and trichloroethylene. Hypomethylation of raf has been associated with enhanced expression of raf in phenobarbital-induced liver tumors of B6C3F1 mice.
The ability to identify possible tumor suppressor genes that are involved with mouse liver tumor development has been hampered by the relative lack of LOH detection in murine liver tumors. It has been speculated that difficulty in detecting LOH in murine liver tumors may be a consequence of the high frequency of tetraploidy in mouse hepatocytes. However, since hypermethylation silencing of tumor suppressor genes is known to occur, this epigenetic mechanism could be involved in murine hepatocarcinogenesis. In addition to altered expression of proto-oncogenes and tumor suppressor genes, mutations and perturbations in other genes such as beta-catenin, E-cadherin, cyclin D1, and EGFR have been documented in mouse liver tumors.
Hormones
With the exception of the female C57BR/cdJ mouse, both spontaneously occurring and treatment induced hepatocellular tumors occur with significantly greater frequency and multiplicity in males than in females.
Castration of 2-day old BALB/c mice followed by gavage administration of N-2-fluorenylacetamide starting at one week of age completely abolished development of hepatocellular carcinomas. Using orchidectomy and ovaiectomy alone as well as with and without subsequent administration of androgens and ovarian hormones or simply administration of hormone without ablative surgery, several studies have shown that testosterone promotes and ovarian hormones suppress development of liver tumors in mice. As an indication of the magnitude of the hormonal effect, neonatal urethane exposure followed by castration or ovariectomy at 6 weeks of age resulted in a 96% and 20% incidence of heptomas in sham-operated male and female B6C3F1 hybrids, respectively, while orchidectomy and ovariectomy hybrids had hepatoma incidences of 62% and 67%, respectively. Using C3H–BALB/c sexually chimeric mice, Tsukamoto and co-workers show that while basic strain differences are genetically determined, male hormonal or micro-environmental factors lead to promotion of liver cancer in both XX and XY hepatocytes.
C57BL/6 × DS F1 mice injected at 18 days of age with 3’-methyl-4-dimethylaminoazobenzene and castrated at 23 days of age had a reduced multiplicity of adenomatous hepatic nodules but castration did not influence the incidence of either adenomatous nodules or carcinomas in males. Ovariectomy of females shortened the latency and increased both the incidence and multiplicity of adenomatous nodules.
Endogenous liver tumor promotion of preneoplastic foci of altered hepatocytes by testosterone is considered a primary explanation for the well documented increased susceptibility of male versus female mice. The tumor promoting effects of testosterone are mediated by the hepatocyte androgen receptor. Castration of male mice leads to a 3-fold increase in hepatocyte androgen receptors, a result that approximates the androgen receptor levels in female mouse hepatocytes.
The greater susceptibility to induction of liver neoplasia in male versus female mice associated with testosterone is attributed to its positive effect on the growth rate of preneoplastic foci of cellular alteration. Either castration of male mice or administration of testosterone to female mice results in a decreased or increased growth rate of preneoplastic lesions, respectively, as well as a corresponding alteration in the multiplicity of liver tumors. Suprisingly, however, the actual testosterone levels among susceptible and resistant inbred mouse strains is not correlated with liver tumor susceptibility. Similarly, the degree of binding of testosterone to hepatocellular androgen receptor is not correlated with the differential liver tumor susceptibility among mouse strains.
In investigating the role of growth hormone in mediating the effects of sex hormones on liver tumor development, investigators at the McArdle lab treated growth hormone deficient C57BL/6J lit/lit mice neonatally with diethylnitrosamine. There was up to a 59-fold increase in tumors in the growth hormone deficient mice versus the wild type C57BL/6J with the effect significantly more dramatic in males than in females. These investigators then bred the growth hormone deficiency onto a C57BR/cdJ and a C3H/HeJ background and demonstrated that growth hormone deficiency suppressed liver tumor development to less than 1%. The authors conclude that growth hormone is a potent endogenous regulator of susceptibility and its absence abrogates the effects of sex hormones and genetic background on liver tumor susceptibility.
Diet and body weight
It has long been know that natural vs synthetic vs semi-synthetic diet plus caloric intake, amino acid composition, lipid content, methyl deficiency, etc. impact safety assessment and hazard identification rodent toxicity and cancer studies. Hancock and Dickie reported 100% incidence of hepatoma in D2CEF1 and CED2F1 hybrids at 8 to 14 months of age when switched to a high protein, high fat diet whereas for the previous 16 years neither hybrid had liver tumors even at 28 to 32 months of age.
In an effort to better control growth, body weight, and age-related disease including tumor incidence, the National Toxicology Program changed from NIH-07 diet to a new diet designated NTP-2000. By reducing the caloric content, mainly by increasing fiber content and reducing protein, the diet could be fed ad libitum. NTP-2000 proved beneficial for a number of variables and, importantly, lowered body weight and the spontaneous incidence of liver tumors in male B6C3F1 mice. An alternative approach of utilizing dietary restriction has been championed by the FDA’s National Center for Toxicologic Research as a means to minimize tumor and survival variability within and between studies and has been shown to inhibit spontaneous and treatment-induced tumorigenesis. A rather extreme dietary restriction of 60% led to increased survival in male and female B6C3F1 mice and resulted in an 8.6-fold reduced liver tumor incidence in control B6C3F1 mice. Dietary restriction started as late as 3 months after treatment with a potent hepatocarcinogen can still have an inhibitory effect on mouse liver tumor development.
There is a strong correlation between body weight at 52 weeks and the subsequent incidence of liver tumors in control B6C3F1 mice and a concern that body weight differences between dosed and control groups could mask carcinogenic effects sensitive to body weight changes. Consequently, reduction of body weight gain would be expected to reduce the spontaneous liver tumor burden. Using tumor risk data from several hundred B6C3F1 mice, Leakey and coworkers constructed body weight weight curves to predict the amount of dietary restriction required to predict a 15 to 20% spontaneous liver tumor incidence. In a chloral hydrate bioassay using this approach, they were successful in achieving their objective and, in addition, the variable feed restriction paradigm provided for a statistically significant dose-response in their study versus the ad libitum treated cohort. As further testimony to the importance of diet, it has been shown that the post-weaning diet in C57BL/6J × Cast/EiJ F1 mice can affect IGFII methylation and lead to permanently decreased IGFII expression via imprinting.
Cell-cell communication
Multiple in vitro and in vivo experimental studies have shown that fully functional gap junctions inhibit both spontaneous and treatment induced neoplasia and that a number of nongenotoxic rodent carcinogens inhibit gap junction cell-cell communication, leading to enhanced cellular proliferation and increased neoplasia. Gap junction cell-cell communication is controlled by connexins which constitute a family of tumor suppressor genes.
For the most part, studies of gap junction intercellular communication in hepatocytes have been carried out on primary cultures. Using primary B6C3F1 hepatocytes, it has been demonstrated that endosulfan and at least one endosulfan metabolite, plus chlordane and heptachlor inhibit gap junctional communication in a dose dependent manner. A dose dependent inhibition of gap junctional cell-cell communication in primary mouse hepatocytes has also been shown for phenoparbital, DDT, and lindane and is most probably mediated via cAMP. Similar inhibition has been documented in primary mouse hepatocytes for monoethylhexylphthalate, tricholoracetic acid, trichloroethylene, nafenopin, and arochlor 1254.
Studies of liver tumor promotion in rats using phenobarbital, polychlorinated biphenyl, and dichlorodiphenyltrichloroethane showed an increase hepatocellular proliferation and decreased gap junction cellular communication, albeit without a quantitative association.
Cell-cell communication can play an important role in mouse hepatocarcinogenesis. Mice deficient in connexin32, the major gap junction protein expressed in hepatocytes, had a 25- and 8-fold increase in spontaneous liver tumors in male and females, respectively, compared to wild-type controls. These same authors showed an increased incidence of liver tumors and a faster growth rate of these tumors in connexin32 deficient mice on a C57BL/6/129/Sv-F1 background compared to controls one year after neonatal treatment with diethylnitrosamine.
While both oncogenes and growth factors have been shown to downregulate gap junctional function and analysis and both rat and mouse hepatic neoplasms have altered gap junctional cell communication, the exact role of cell-cell communication in the sequential development of mouse hepatocarcinogenesis is unclear. However, hypermethylation inactivates connexin genes, suggesting that methylation may contribute to carcinogenesis by disruption of gap junctional intercellular communication.
Viral infection
A retrospective review of over 30 control groups and over 30 low dose and high dose groups of B6C3F1 mice of each sex was undertaken to determine the effect of viral infection on tumor incidence in conventional cancer bioassays. Sendai virus infection was associated with increased incidence of liver tumors and lympomas in B6C3F1 males but no increase of tumors in female B6C3F1 mice. The increased tumor response may be a consequence of higher survival of control, low-does and high-dose groups.
Reversibility (Conditional Hepatocarcinogens)
It is certainly well documented that a major hallmark of liver tumor promotion by nongenotoxic rodent hepatocarcinogens is the regression of preneoplastic foci and nodules following cessation of treatment. A dramatic example of the extent of this process is the regression of 30% of chlordane induced hepatocellular adenomas and carcinomas within a few weeks after stopping treatment. Regression of frank liver neoplasia has been reported in humans following cessation of growth hormone supplements containing androgens, in women after ceasing use of oral contraceptives, and in rats after cessation of nafenopin, phenobarbital and clofibrate, and the peroxisome proliferators WY-14,643. Agents which require continual administration for the stable presence and growth of preneoplastic and neoplastic rodent liver lesions are probably best categorized as conditional hepatocarcinogens.
Historical Controls
For assessment of liver tumor data in cancer bioassays, laboratory specific historical control data are extremely valuable in putting unusual high or low tumor responses into perspective. Acquiring accurate historical control data for liver tumors of different inbred mouse strains is problematic because of a host of variables such as caging, diet, individual study duration, route of test article administration, and the period of time over which the control data are acquired. The best data come from labs with consistent study protocols and with periodic updating of a moving window of observation to generate the most relevant contemporary historical control information. While historical control data from organizations such as the National Toxicology Program and the FDA National Center for Toxicological Research are readily available, similar data from industrial organizations is generally not available to the public. Based on extraction of concurrent control data from published reports, some crude estimates could be generated but these would constitute isolated examples of concurrent controls rather than solidly reliable historical controls. Based on web site data for March 2007, the NTP mean (± SD) historical control data for B6C3F1 mouse liver tumors with all routes combined and using NTP-2000 diet is shown in Table 3.

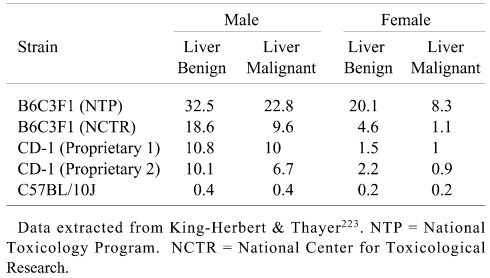
Some comparative historical data extracted from the NTP Workshop on liver tumors in different mice but without background husbandry and study duration data are presented in Table 4. There are dramatic strain and stock differences in liver tumor incidences and, based on data in Tables 3 and and4, the NTP B6C3F1 mouse has a particularly high and variable background incidence of liver tumors.
Rationale for Selection of Experimental Mice
Strain and stock selection of experimental mice for toxicity and carcinogenicity studies is based partly on ready accessibility, organizational history, and the specific purposes of the study. Unfortunately, the reasons for selection of specific mice for cancer bioassays are often obscure. However, once selected for use in hazard identification screening, there are two primary reasons for the continued use of the particular strain: (1) reluctance to lose the historic control database, and (2) historic inertia. In the early phases of the National Cancer Institute cancer bioassay program several mouse strains including HaM/ ICR, CD-1, and strain A were used. The report of a large scale study involving 20,000 mice exposed to 127 different pesticides and industrial chemicals compared responses from two F1 hybrids: (C57BL/6 × C3H/Anf) F1 and C57BL/6 × AKR)F1. The B6C3F1 hybrid was more effective at identifying the chemicals that were considered known carcinogens and, as a result, it ultimately became the default mouse for the National Cancer Institute cancer bioassay program. Use of this hybrid has continued with the transfer of the bioassay program to the National Toxicology Program.
A recent National Toxicology Program workshop was held to consider the most appropriate rodent strains for hazard identification studies and, although there was considerable discussion considering mouse strains other than the B6C3F1, a decision to change has not been forthcoming. The contemporary National Toxicology Program philosophy would consider use of either additional or alternative mouse strains for specific studies on a knowledge-based approach to testing and based on specific issues related to the agent under study. There was general interest, however, in considering use of multiple isogenic mouse strains, especially if appropriate background information were available and in spite of potential logistical constraints.
The outbred CD1 and NMRI stocks and inbred C57BL/ 10 mice are generally favored for carcinogenicity studies by the pharmaceutical, pesticide, and agrochemical communities. A comfort level in use of these particular mice plus a robust historical control database are reasons these organizations maintain use of their preferred mouse.
Evaluation, Interpretation, and Relevance
General
As a separate exercise from assessing the relevancy of a positive or negative cancer bioassay response, it must first be determined if the response observed in the test animals is credible. A negative cancer outcome in both sexes of two test species is generally interpreted to reflect the probability that the agent under test is not a potential human carcinogen. This judgment is not made in a vacuum but rather takes into account many other factors including whether the agent is genotoxic, the toxicokinetics in the bioassay models, evidence of a biological response indicative of adequate exposure, sufficient survival for the duration of the bioassay, etc. Given that all factors favorably support the bioassay outcome, it is probable that existing or pending regulatory actions will be mitigated. However, it is readily understood that one can never prove a negative; susceptible human subpopulations or individuals might react unfavorably to agent exposure; and the bioassay outcome could be reflective of a false negative response.
Multi-site and trans-species carcinogens
Positive cancer responses in the bioassay test species engender a spectrum of interpretative response and sometimes a considerable degree of controversy. This follows from the fact that bioassays yield varying degrees of response. At one end of the spectrum is the clear response in which multiple cancers occur early in the study involving multiple tissues in both male and female rats and mice – a multi-site, trans-species rodent carcinogen. At the other end of the spectrum of response is the situation where there is a marginal increase in a common spontaneous tumor and the response is seen in one sex of one species and typically only at the highest dose. For those individuals who subscribe to a conservative public health policy, these responses are often considered indicative of potential human risk for developing cancer, at the very least for susceptible human subpopulations or in situations where there may be in uterohuman exposure. The majority of positive outcomes, which in the case of the National Toxicology Program represents approximately 50% of agents tested, fall between the extremes of response and are generally classified as showing either clear or some evidence of carcinogenicity. Appropriate regulatory agencies and bodies then take the bioassay outcome into account as one of the factors in the weight of evidence for determining the potential human health hazard. Multi-site and trans-species carcinogens would provide more compelling justification for protective regulatory decisions than a single sex, single species marginal response. Situations where there is an equivocal response ideally require additional, and perhaps more rigorous, studies; but cost considerations generally preclude additional studies except in unusual circumstances.
Mouse liver carcinogens
Concern about a mouse liver tumor response comes about in situations where it is the sole response in a two-species carcinogenicity study, is typically seen in liver tumor susceptible strains when given high doses of nongenotoxic agents, when the likely human exposure is significantly lower than bioassay responsive doses, when the likely mode of action is expected to have a threshold, and when the mode of action is not relevant to humans. Given what we have learned from various bioassay toxicity and carcinogenicity databases generated over the past 40 years, short and medium term exposures can reasonably be expected to identify predictive rodent- and chemical-specific biomarkers and, thus, to establish predictive testing strategies. This may be relatively easy for prediction of liver responses by use of clinical chemisty, organ weight, histopathology, and cell proliferation measurements and should work equally well for rats as well as mice.
Mouse debate
Over the last three decades and especially during the last few years, there has been considerable concern and debate regarding the utility of the mouse for long-term rodent carcinogenicity testing. A few quotes (without attribution) serve to illustrate one position regarding the utility of mouse bioassays:
“It is not appropriate to make human risk assessment decisions based on a mouse liver tumor response”
“…. it is suggested that strong consideration be given to deleting the mouse as a routine test animal ….”
“So, my conclusion is, in the future, probably the near future, with sufficient scientific evidence added, we can eliminate the long-term mouse bioassay from our protocol.”
“For non-genotoxic hepatic tumor promoters, the weight of the evidence would indicate that a mouse liver tumor response is not a relevant indication of human cancer risk.”
“…. have strengthened my views that from a regulatory standpoint the use of mouse strains with a high spontaneous incidence of hepatic tumours for routine carcinogenicity testing is undesirable.”
“The utility of the mouse for purposes of routine screening of chemicals for carcinogenic potential is, therefore, highly questionable.”
A primary basis for recommendations that the mouse no longer be used for carcinogen hazard identification stems from the liver tumor response. Some argue that, at least for pharmaceuticals, the mouse cancer bioassay is redundant in that it has not provided evidence of carcinogenicity that was not already identified in the rat cancer bioassay. The proponents of using the mouse bioassay are concerned that dropping it from the armamentarium will preclude the possibility of identify trans-species carcinogens. The current development of new genetically engineered mouse models to better understand and combat cancer and ongoing efforts to map the genome of multiple inbred mouse strains suggest that it would be prudent to retain the mouse as a cancer bioassay species. Although the debate regarding the utility of the mouse as a cancer bioassay model will probably continue, it appears that mice will retain a role in understanding the complexity of cancer as a disease, identifying causative factors, serving as a screening model for hazard identification, and providing a basis for therapy and, hopefully, prevention.
Relevance
Of all the concerns and controversies surrounding rodent cancer bioassay programs, relevance to potential human disease is of critical importance. Since the objective of the rodent cancer bioassay is to provide information that will permit the avoidance, reduction, and prevention of carcinogenic risk to humans, the relevance of the bioassay must be defined. Not surprisingly, there are two schools of thought on this topic. It is generally accepted that agents whose mode of action involves direct interaction with and alteration of DNA should be considered to have human carcinogenic potential. These DNA reactive agents are typically considered to not have a threshold, although there may be some low level of exposure to genotoxic agents that simply will not result in cancer in a human lifetime. The majority of agents tested in contemporary rodent cancer bioassays, however, are non-genotoxic and, if they indeed do lead to cancer in rats and/or mice, the mechanism by which that rodent cancer occurs should ideally have relevance to humans. Over the years, diligent investigative work subsequent to observed cancer responses in the rodent bioassay, has provided convincing evidence that some bioassay cancer response are rodent-specific and simply not germane to humans. Agencies have tended to mitigate regulatory decisions when clear rodent-specific mode of action data are provided and all other factors are considered, including a cost-benefit analysis.
The Future
The rodent cancer bioassay is unlikely to be abandoned in the near future. Even given an appealing alternative for identifying carcinogenic hazard, just the process of validating that alternative and overcoming historical inertia could easily take several years. Furthermore, the rodent cancer bioassay has evolved to address various toxicities other than just cancer. Concerns about reproductive toxicity, neurotoxicity, immunotoxicity, and developmental effects have captured public awareness and organizations are increasingly addressing the issue of toxicity and other non-cancer consequences for human health.
Contemporary rodent bioassay design frequently incorporates sub-studies for the generation of information about mode of action. Concerns about the sensitivity of the rodent bioassay and growing concern about long-term and trans-generational human health issues are leading to the prospect of running bioassays that commence with in utero exposure followed by continuation of exposure during nursing and following weaning. This development will pose considerable logistical considerations for testing as well as being resource intensive. The general belief is that in utero exposures will provide a more sensitive bioassay animal model. Its use will undoubtedly identify more agents as hazards, including cancer hazards. If such studies are undertaken they will hopefully be designed to simultaneously develop predictive biomarkers to help obviate the need for continual long-term resource intensive testing.
While the majority of bioassays have been carried out on single specific agents in the workplace and general environment, there is a growing interest in understanding the health consequences for the total environmental load of exposures and in testing relevant mixtures of agents that would likely reflect realistic human exposures. Examples include complex non-standardized agents such as readily available over-the-counter herbal medicines, combinations of exposures from water disinfection byproducts, physical exposures from electromagnetic fields and cell phones, nanoscale materials, and endocrine disruptors to name a few. This type of testing will require novel exposure scenarios and will create logistical hurdles and new issues of interpretation and relevance. The problems will be all the more complex as the rodent bioassay is expanded to include non-cancer endpoints, exploration of interacting modes of action, and identification of biomarkers of exposure and effect that might have applicability in protecting human health.
Footnotes
* The contents of this review were presented as a background paper for the Standing Committee on Risk Analysis Issues and Reviews, National Academy of Sciences, in conjunction with a workshop entitled “Mouse Liver Tumors: Benefits and Constraints on Use in Human Health Risk Assessment, Qualitative and Quantitative Aspects” held on November 8-9, 2007 in Washington, DC.